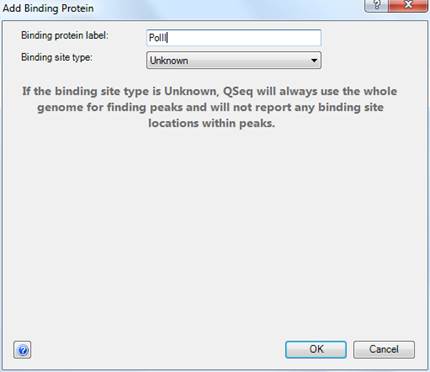
The Add Binding Protein dialog allows you to define binding proteins and binding sites for your experiment. To create a new binding protein, type in a name in the Binding Protein Label field and choose a binding site type from the available options:
•Unknown – QSeq will use the whole genome for finding peaks and will not report binding site locations within peaks.
•Transcription Factor Database – Choosing this option allows you to select a binding site pattern from DNASTAR’s transcription factor database. Select the organism and the name of the binding site and transcription factor from the respective drop-down menus. If you have already entered a label for the binding protein, QSeq will automatically select the transcription factor from the list that matches this name when possible. Likewise, if you make a selection in the transcription factor drop-down before filling in the transcription factor name, QSeq will fill in the transcription factor name to match your selection. The Sequence field shows the IUPAC nucleic acid codes for the selected pattern. You may wish to click on the Summary info link for more information about the currently selected pattern.

•Type-in Pattern – Choose this option if you prefer to specify your own binding site pattern for your binding protein. QSeq recognizes IUPAC nucleic acid and regular expression syntax for these patterns. A key to the syntax is provided within the dialog.

•Position Weight Matrix – If this binding site type is selected, QSeq will use a position weight matrix to locate binding sites. Select Use JASPAR position weight matrix then choose the species and factor names from the drop-down menus to use one of the matrices that is shipped with ArrayStar. Or select Load position weight matrix from file and then Select Matrix file to browse to and load a custom matrix file.
QSeq will calculate the log-odds for each sequence given the selected matrix. The score for a single character at a particular position in the matrix is equal to the log2 of the likelihood of seeing that character at that position in the data used to generate the matrix divided by the background likelihood of seeing that character at that position.
For example, if the matrix is derived from 80 sequences and in 70 of those sequences there is an “A” in position 1, the log odds score of seeing the character “A” in position 1 is log2((70/80)/(20/80)) = 1.80. If a “C” occurs 1 time in position 1 of the training sequences, the log odds score of seeing the character “C” in position 1 is log2((1/80)/(20/80)) = -4.32. To get the log odds score for the whole sequence, QSeq sums the log odds scores of each character in the sequence.
A sequence is considered to "match" the matrix if its score is greater than or equal to the specified Threshold. By default, the threshold value is half of the average of the log-odds scores of sequences that were used to train the pattern. You can increase the threshold for more stringency or decrease it for more matches.
