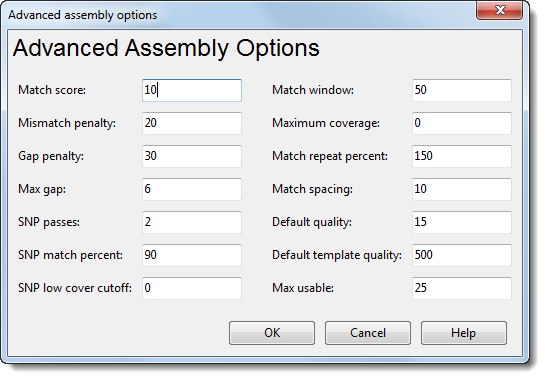
In a de novo or special reference-guided workflow, you can open the Advanced Assembly Options dialog by pressing the Advanced Assembly Options button from the Assembly Options dialog.
Default parameters vary according to the sequencing technology and project type specified elsewhere in the wizard, and values seldom need to be changed. If desired, edit values for:
•Match score – The score for a base match during an alignment. This score contributes to the pairwise score used to calculate match percentage. Increasing this value will allow for longer or more frequent gaps, thus forcing bases that match to be assembled together.
•Mismatch penalty – The penalty for a base mismatch during an alignment. This penalty is deducted from the pairwise score used to calculate match percentage.
•Gap penalty – The penalty for opening or extending a gap during an alignment. This penalty is deducted from the pairwise score used to calculate match percentage. A high gap penalty suppresses gapping, while a low value promotes gapping.
•Max gap – The maximum number of gaps allowed per 1000 bases in the alignment.
•SNP passes – The number of times SeqMan NGen will cycle through a templated assembly, attempting to fill in regions with zero or low coverage due to SNPs.
•SNP match percent – The minimum match percentage required during passes to fill in SNP regions. The default value will change depending on the type of assembly and the read technology you selected.
•SNP low cover cutoff – The minimum coverage required in an assembly to be excluded from SNP passes. SeqMan NGen will include regions in an assembly that have coverage less than the value specified as well as regions with zero coverage when it performs SNP passes. (See the SNP passes parameter above.)
•Match window – The size of the window used to calculate match percentage.
•Maximum coverage – The maximum depth of coverage allowed in a templated assembly. SeqMan NGen will not exceed the coverage specified by this threshold. The default value of “0” equals unlimited coverage.
Note: This parameter is only available for templated assemblies, and should be used with caution, as it will limit the number of sequences included in the assembly.
•Match repeat percent – The percent frequency a mer occurs compared to its expected frequency. Mers exceeding this value are flagged as repeated and not used as mer tags in determining overlaps.
•Match spacing – The length of the window of a sequence read where at least one mer tag will be chosen. The default value will change depending on the read technology you selected.
•Default quality – The value used for the base quality of sequences without quality scores.
•Default template quality – The value used for the base quality of template sequences without quality scores.
•Max usable – Any mers occurring more frequently than the Repeat Handling expected coverage value multiplied by this value are disregarded as mer tags from the assembly.
Once you are finished, click OK to save changes and return to the Assembly Options dialog, or Cancel to return without saving changes.