Clicking the Advanced (Assembly) Options button from certain versions of the Assembly Options dialog launches a multi-tabbed Advanced Assembly Options dialog. This help topic describes options available in the Peak Detection tab.
The Peak Detection tab has a different appearance depending on the peak detection method chosen in the Assembly and Signal Processing screen: MACS, ERANGE2, or ERANGE3. Default parameters for each version are optimized based on other selections you made in the wizard. Because of this, values seldom need to be changed.
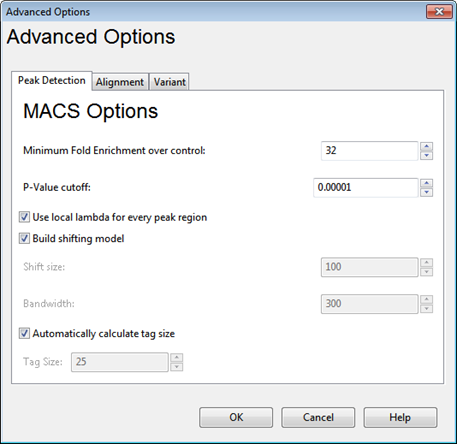
MACS Options:
•Minimum Fold Enrichment over control – This parameter controls how enriched a peak must be, compared to the background read distribution, in order to be considered in building the peak model. If a previous assembly attempt returned the error message “too few paired peaks to build a model,” we recommend using a lower number in this box.
•P-Value cutoff –Local read distribution is compared to the threshold value entered here in order to calculate whether a peak should be counted as “real.” SeqMan NGen calculates the likelihood that a detected peak is actually a peak based on the local read distribution and only returns peaks with values below the P-Value cutoff. The default P-value cutoff is 0.0001. This doesn’t mean that values greater than 10-5 are filtered out, but rather that they are not included in the count of “real” peaks.
•Use local lambda for every peak region – Lambda is a parameter used to define a Poisson distribution which MACS uses to determine the expected number of reads in a given region. When this option is checked, the Poisson distribution is calculated for the peak region and for three regions surrounding the peak. MACS can use this information to determine the expected number of reads in a given region. If the option is unchecked, then the local distributions are not calculated. Instead the expected distribution is based on the total number of reads and the effective size of the genome.
•Build shifting model – Check this option to have MACS build a model based on the data to determine the width of and the distance between the "paired peaks.” Alternatively, leave this option unchecked to set Shift Size and Bandwidth values manually. The Shift Size is the distance each of the paired peaks will be shifted to try to center them over the actual binding site. The Bandwidth value defines the expected width of peaks. SeqMan NGen will search for peaks using a window twice as long as the bandwidth.
Note: The creators of the MACS algorithm advise disabling model building when dealing with broad peaks (i.e. binding sites); for example, when studying histone binding.
•Automatically calculate tag size – MACS treats all reads as though they have equal length. To explicitly specify that length, check Automatically calculate tag size and enter a length in the Tag Size text box.
ERANGE2 Options:
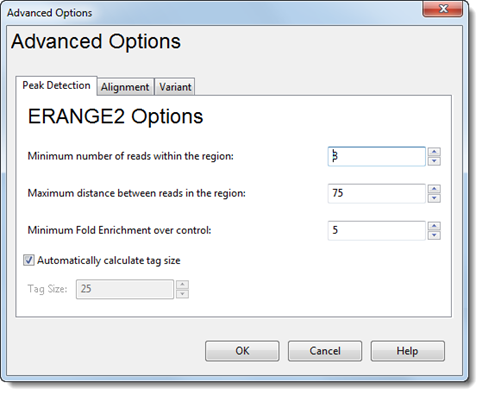
•Minimum number of reads within the region – Type in the desired minimum threshold.
•Maximum distance between reads in the region – Type in the desired maximum distance.
•Minimum Fold Enrichment over control – This parameter controls how enriched a peak must be, compared to the background read distribution, in order to be considered in building the peak model. If a previous assembly attempt returned the error message “too few paired peaks to build a model,” we recommend using a lower number in this box.
ERANGE3 Options:
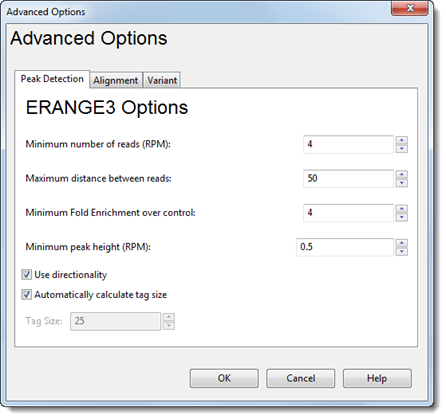
•Minimum number of reads (RPM) – Type in the desired minimum number of reads.
•Maximum distance between reads – Type in the desired maximum distance.
•Minimum Fold Enrichment over control – This parameter controls how enriched a peak must be, compared to the background read distribution, in order to be considered in building the peak model. If a previous assembly attempt returned the error message “too few paired peaks to build a model,” we recommend using a lower number in this box.
•Minimum peak height (RPM) – Type in the minimum peak height threshold.
•Use directionality – If this box is checked, SeqMan NGen will consider the proportions of forward and backward reads found in each peak. If the peak does not meet an internal threshold requirement for directionality, it will be discarded. Checking this box ensures that the evidence for a peak is balanced between the strands.
•Automatically calculate tag size –ERANGE3 treats all reads as though they have equal length. To explicitly specify that length, check Automatically calculate tag size and enter a length in the Tag Size text box.
**************************
When you have made the desired selections in this tab of the Assembly Options dialog, click another available tab to make changes there. If you don’t need to make further changes, click OK to close the Advanced Assembly Options dialog and return to the Assembly and Signal Processing or Assembly Options screen.