Reference-guided assembly with gap closure is a semi-automated assembly option that utilizes both reference-guided (“templated”) and de novo assembly steps to resolve three types of structural variation (SV): insertions, deletions and replacements (indels) with minimal user intervention.
To follow this workflow, choose Whole Genome in the Choose Assembly Workflow screen, and Reference-guided assembly with gap closure in the Choose Assembly Type screen.
Your data should be from a haploid genome with at least one mate pair data set with read lengths of 100 bases or greater. Your total number of reads should be 10 million or less. If you use a larger data set, only the first 10 million reads will be used. For mate pair data, equal numbers of matching forward and reverse reads are processed.
Steps 1-3c: Assembly in SeqMan NGen
During assembly, data is processed in several stages (see figures below):
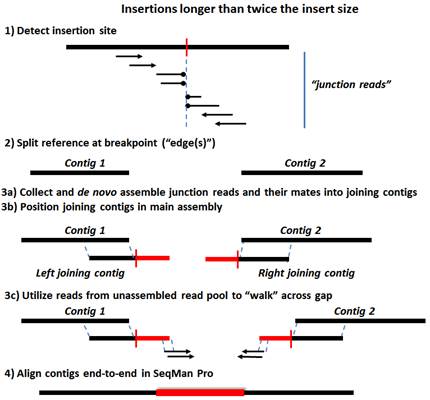
Step 1) Data is mapped and aligned to a user-defined reference genome and then analyzed for characteristic SV motifs.
Step 2) The reference sequence is split at the detected SV sites, forming a series of ordered contigs.
Step 3a) Mate pair and split reads from each SV event are collected in site-specific pools and assembled de novo. Deletions are detected using three types of data: split reads, spanning paired-end reads, and sequence coverage information. For insertions and replacements, mate pair reads corresponding to the new sequence are collected from the unassembled read pool. Only reads anchored by mates flanking the SV in the main assembly are used at this stage.
Step 3b) The de novo assembled contigs are then brought into the main assembly and positioned consistently with the mate pair information.
Step 3c) For SVs where the gap is not completely covered by the de novo assembled contigs (e.g. insertions longer than twice the size of the insert library), additional reads from the unassembled read pool matching and extending the ends of the joining contigs are added in an attempt to “walk” across the gap. This walk is terminated when either no new reads are found or when a repeated element is encountered.
At the end of assembly in SeqMan NGen, two types of output files are produced. These allow the project to be evaluated and further processed in SeqMan Pro:
•An .assembly package with a non-editable BAM formatted alignment file of the initial reference-guided assembly without further processing.
•The fully processed assembly in an editable SeqMan .sqd file format.
Step 4: Further Processing in SeqMan Pro
Step 4) The editable .sqd document, containing the fully processed assembly, is used for gap closure to complete the new sequence.
In SeqMan Pro's Project Summary window, the contigs will appear in a single ordered scaffold with the de novo generated contigs. The values in the contig position column of the window roughly correspond to the 5’ position of the first base of that contig in the reference genome, although the position is generally shifted 20 bp downstream to accommodate positions for the gap filling contigs. The ordered contigs can be merged using SeqMan Pro's Contig > Align Contigs End-to-End option. This option ensures that only adjacent contigs are considered for merging, mitigating against false joins caused by repeat elements. With sufficient depth of coverage, this step should close a significant number of gaps. However, some gaps may remain. These may be caused, for example, by long insertions that could not be reliably walked across in this automated fashion. The remaining gaps that require more manual intervention can be closed using the suite of tools in SeqMan NGen and SeqMan Pro.
Evaluation in SeqMan Pro
The .assembly package allows you to inspect the initial reference-guided assembly and detected SV events via SeqMan Pro's Structural Variation table. Single nucleotide polymorphisms (SNPs) and small insertions and deletions can also be inspected using the SNP table.
The following images show Steps 1-4 for deletions and three types of insertions. The terms “split reads” and “junction reads” are used in some of these images and are defined as:
•Split reads – Reads in which the first portion matches one location in the genome, and the adjacent portion matches a downstream location on the same strand. The endpoint of the first segment and the start point of the second segment generally define the breakpoints of the deletion, although the exact positions may vary by a few bases in some cases. The presence of multiple “split reads” at a given position is required to avoid spurious splits caused by, for example, micro-repeats in the genome.
•Junction reads – Mate pair reads where one read aligns either upstream or downstream of the structural variant, and its mate aligns either on the other side (“spanning pairs”) or within the new sequence (in the case of insertions and indels). In the latter case, reads within the inserted sequence are identified from the unassembled read pool by virtue of their mates in the assembly.