In the Input Reference Sequences screen, the following options are available for the templated Transcriptome/RNA-Seq workflow only.
•The Add .Transcriptome button is only available in this workflow, and is used to add .transcriptome packages.
•Before (and usually after) adding files, the following options are available near the bottom of the screen:
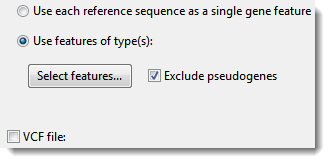
o Use each reference sequence as a single gene feature – Select this option to use each entire template sequence as a target for mapping reads. When this box is checked, RNA-Seq workflows treat these sequences as isoforms rather than genes. Comparing the mapping coverage to related isoforms provides a way to investigate differential expression of alternately spliced transcripts.
o Use features of type(s) – Select this option to only report results when a specific type of feature is used as the target for mapping reads. (If your template sequences do not contain features, this option will be disabled.) Note that mapping occurs regardless of the type of feature annotation. However, when you choose this option, the mapping results for unwanted feature types will not be reported. Use the Select features button to specify which feature types to use. In the ensuing dialog, the right pane shows features that will be used as targets, and the left pane shows other available feature types.
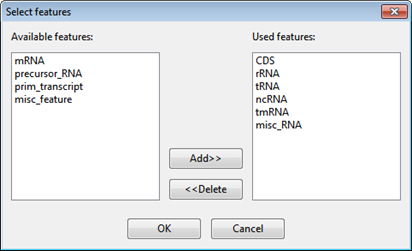
Select items and then use the Add>> and <<Delete buttons to populate the two sides such that only the desired feature types remain in the right-hand pane. Click OK to return to the Input Reference Sequences screen.
o Exclude pseudogenes –As with the previous option, mapping occurs regardless of the type of feature annotation. However, when you choose this option, mapping results for features with /pseudo in their annotations will not be reported.
o VCF file – For information on uploading a VCF file, see Adding and Removing Accessory Files (VCF, BED, etc.).
•If you use the Add Folder button to add a .transcriptome package (see Using Transcript Annotation (StarBlast) Workflow Output as the Template), the options above are replaced with the following:
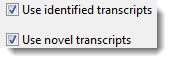
Check one or both boxes to Use identified transcripts, Use novel transcripts, or both. Only the .fas file(s) containing the selected type(s) of transcripts will be imported into the project.