In the past decade, the ability to determine complex mechanisms underlying disease has been made easier by a variety of factors, especially the availability of large amounts of data. In fact, with the continuously decreasing costs of obtaining whole exome DNA sequence data through next-generation sequencing (NGS) technologies, the challenge becomes less about the available data and more about the ability of researchers to tease out meaningful correlations.
Today, researchers often find they must wait for the limited availability of biostatisticians and bioinformatics teams. However, with the right tools, it is hoped that the solutions to Mendelian and complex diseases will be discovered by investigators at their own desktop computers.
DNASTAR combines the computational power required to prioritize relevant factors and visualize correlations in an easy-to-use, integrated software pipeline that puts the power of association studies into the hands of clinical researchers.
A Powerful Integrated Software Pipeline
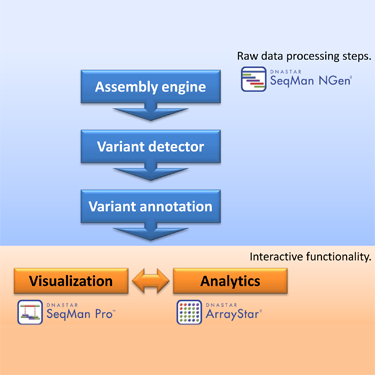 The number and size of NGS data sets that are needed to conduct association studies can pose some challenges. First, the massive amount of raw data (typically 10-30 GB for a single exome and 300-400 GB for a whole genome) requires substantial computer resources for processing as well as for storage and management. Second, there is a series of computational tools required:
The number and size of NGS data sets that are needed to conduct association studies can pose some challenges. First, the massive amount of raw data (typically 10-30 GB for a single exome and 300-400 GB for a whole genome) requires substantial computer resources for processing as well as for storage and management. Second, there is a series of computational tools required:
1) A large capacity NGS reference-guided assembler
2) A variation detection module
3) A variant annotation module
4) A visualization package for inspecting alignments and variant calls
5) An analytics module for comparing variants across samples including statistical analyses and discrete filtering
Stringing together and running the software tools needed to accomplish these tasks can be quite a hurdle. But, with the integrated Lasergene Genomics Suite, the flow of data is facilitated with an easy-to-use, intuitive, graphical interface. The suite consists of three programs: SeqMan NGen, SeqMan Pro, and ArrayStar.
 Case Study: Kabuki Syndrome
Case Study: Kabuki Syndrome
As a demonstration of DNASTAR’s pipeline, a rare Mendelian disorder known as Kabuki syndrome was used. Exome data sets were obtained through dbGaP. These data sets were from the published Kabuki syndrome study (Ng et. al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 42, 30-35 (2010)). The syndrome, which is caused by autosomal dominant mutations, is rare with approximately 400 cases reported worldwide.
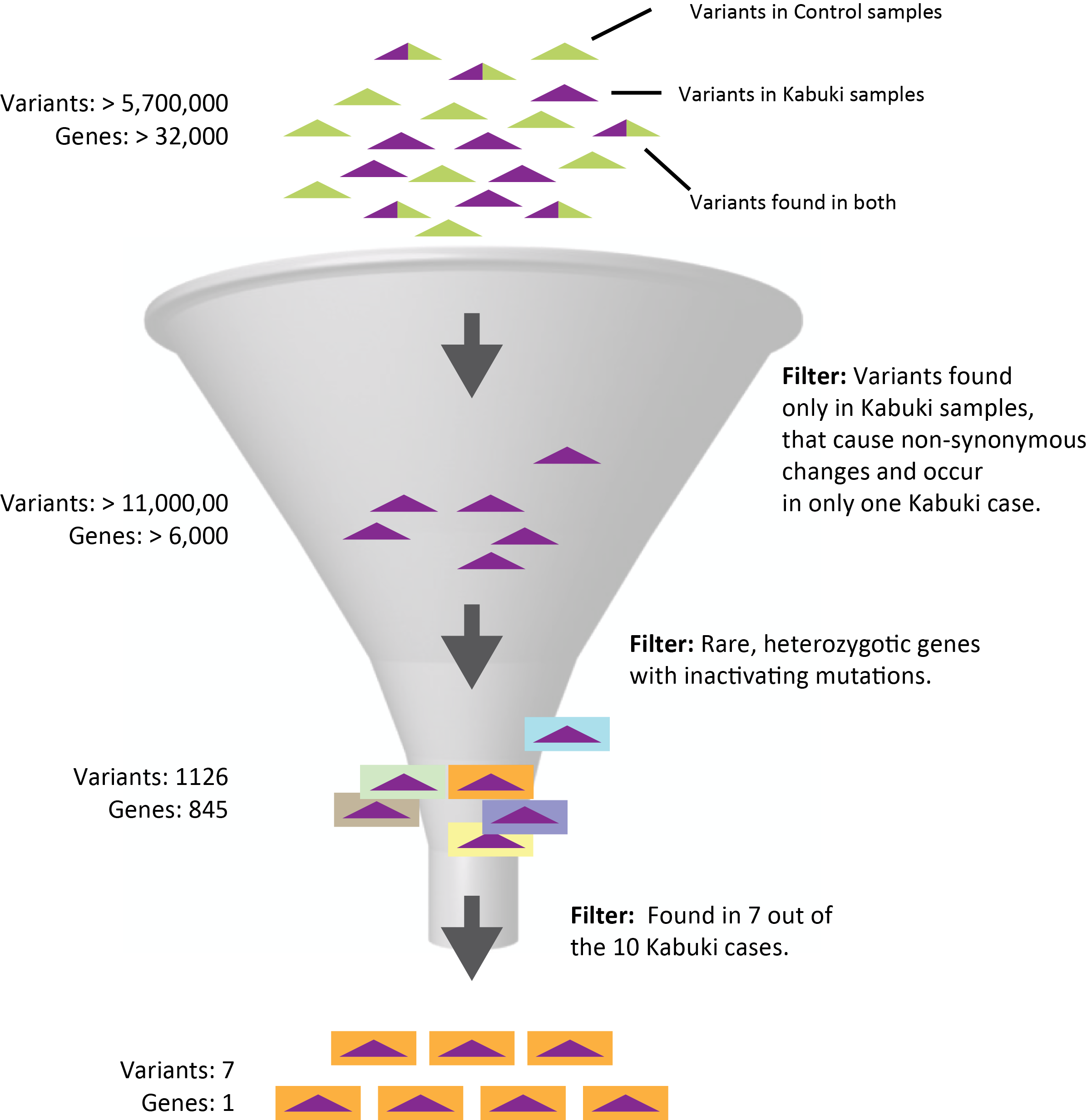
Ten case and eight control exome data sets were independently aligned to the human genome reference sequence using SeqMan NGen which also identified and annotated variants. Variants from each assembly were then loaded together into ArrayStar resulting in over 5.7 million independent positions located in about 32,000 genes across all samples after coalescing. The samples were then organized into two groups, Kabuki and Control, to facilitate subsequent filtering.
We first filtered at the variant level making three assumptions based on knowledge of the disease: 1) causal mutations would be non-synonymous changes, 2) causal mutations arose de novo so variants would occur in only one case sample and 3) no control sample would have any of the mutations. Stringent quality metric thresholds were also imposed to reduce noise. Over 11,000 variants in 6,352 genes met the criteria and were saved as a “SNP set.”
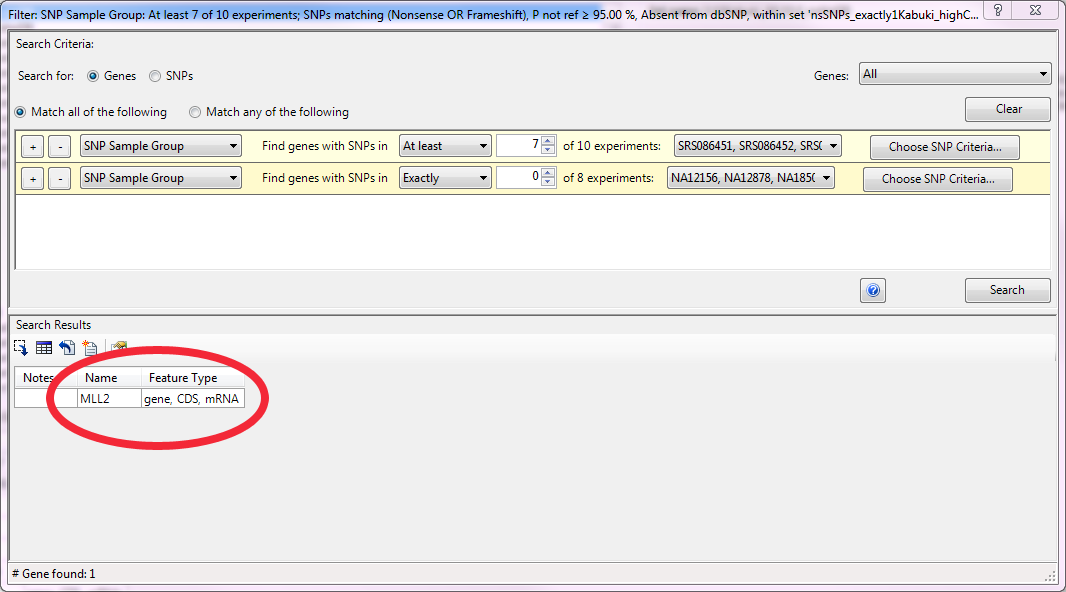
This SNP set was then used as the variant pool in a second filtering step. This time to identify genes with mutations that met the following criteria: 1) mutations were inactivating (nonsense or frameshift), 2) they were rare and therefore not in dbSNP and 3) they were dominant and therefore occurred as heterozygotes. 845 genes met those criteria in at least one case sample. However, by increasing the level of detectance to 7 of 10 case samples the number of candidates was reduced to one, MLL2, consistent with the results of Ng et. al.
The DNASTAR software makes this type of filtering easy for researchers with an intuitive filtering interface:
Conclusions
Exome sequencing has revolutionized our ability to detect common, rare and private variants in the coding genes of an individual. By sequencing case and control cohorts and then comparing across the spectrum of variants, the genetic causes of Mendelian and complex diseases are being uncovered. NGS technologies and facile software pipelines that integrate assembly, variant calling/annotation and association analyses are essential partners in this endeavor.

"/><text x="50%" y="50%" fill="red" text-anchor="middle" dominant-baseline="middle">%26%23x26A0%3B︎</text></svg>)

Leave a Reply
Your email is safe with us.