
The Analysis Options wizard screen is the third of five screens used to set up a gene homology alignment. This screen allows you to customize options related to post-assembly analysis.
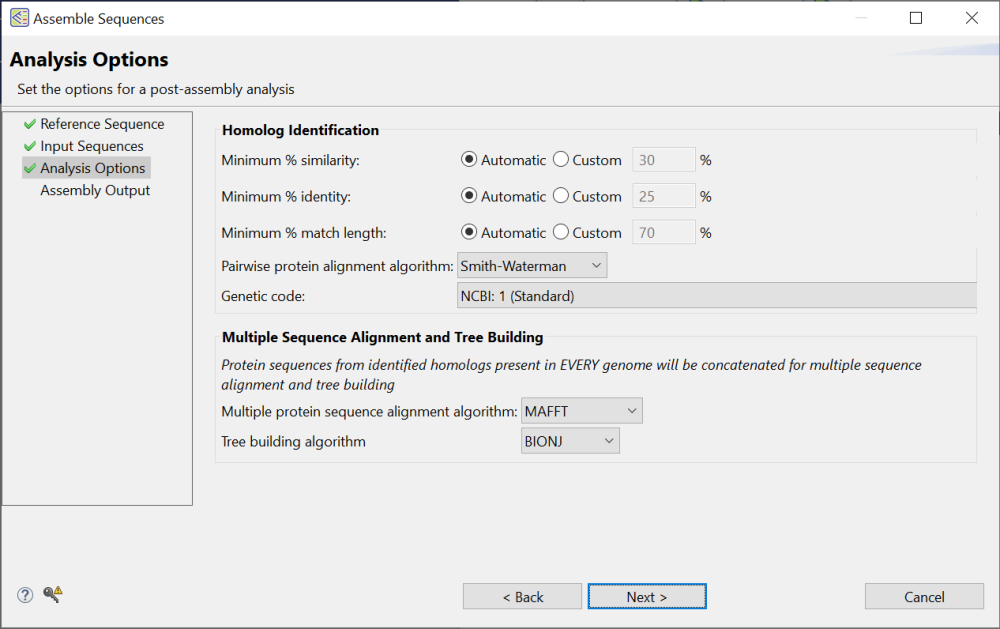
You can leave all options at their default values or make custom selections, as desired. Each option is described below:
- Minimum % similarity – The minimum percentage of relatedness between two aligned sequences. Select Automatic to use the default value of 30% or Custom to enter your own value.
- Minimum % identity – The minimum percentage of bases that must match exactly when any two sequences are aligned. (NCBI definition). Select Automatic to use the default value of 25% or Custom to enter your own value. For example, if you wanted no more than 25 mismatches for a sequence with a length of 500. You would enter 95% (475/500).
- Minimum % match length – The minimum percent of the gene homolog’s length that must align with the reference. Select Automatic to use the default value of 70% or Custom to enter your own value.
- Pairwise protein alignment algorithm – Select Smith-Waterman or Needleman Wunch. These methods are described in Choosing a pairwise alignment method.
- Genetic code – Choose from over two dozen genetic codes, including codes from NCBI and DNASTAR Lasergene.
- Multiple protein sequence alignment algorithm – Choose from MAFFT, MUSCLE, Clustal Omega, or ClustalW. These methods are discussed in Multiple alignment methods and options.
- Tree building algorithm – Select BIONJ, RAxML, or RAxML-NG. These methods are described in Generate a phylogenetic tree.
When you are finished making selections on this screen, click Next to proceed to the Assembly Output screen.
Need more help with this?
Contact DNASTAR