
The Pairwise view displays sequences after performing a pairwise alignment or a BLAST or Entrez database search. Sequence names selected in this view are simultaneously selected all other views in MegAlign Pro. Similarly, sequence names selected elsewhere will be highlighted in the Pairwise view.
To access the Pairwise view, do either of the following:
- Perform a pairwise alignment. In this case, the Pairwise view opens automatically to display the results. To open the view at a later time, click on the Pairwise tab or choose View > Pairwise > Show. If there are multiple Pairwise tabs, hover over each tab to see a tooltip showing which sequences were involved in that alignment.
- Perform a BLAST search. In this case the Text, Table and Pairwise views are displayed together in a single popup window that is separate from the main MegAlign Pro window. To reopen the Pairwise view (and the associated views) at a later time, click on the Status link in the Jobs panel.
In the left of the header, the Align (left) drop-down menu is used to specify the target sequence, which should be the longest sequence of the two. The with (right) drop-down menu is used to specify the query sequence. Specifying a different sequence in either menu will initiate a new pairwise alignment; the original pairwise alignment and its statistics will be replaced with information from the new alignment.
Summary area:
The summary area describes the alignment engine and settings that were used to create the pairwise alignment.
If you aligned a short sequence to a chromosome-length sequence using the Align to Chromosome: DNASTAR method, the area will look like the image below.
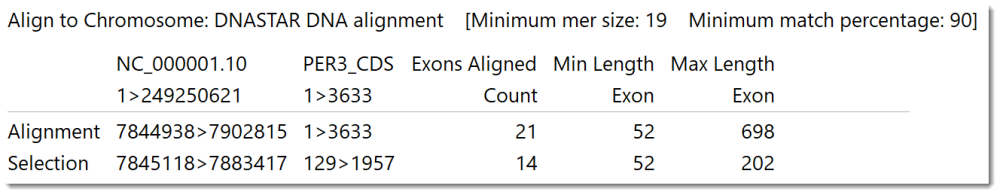
The columns will include the following information:
| Value | Description |
|---|---|
| Exons Aligned Count | The number of exons in the short sequence that MegAlign Pro was able to align to the chromosome while honoring the Minimum mer size and Minimum match percentage thresholds. |
| Min Length Exon | The length of the shortest exon in the alignment. |
| Max Length Exon | The length of the longest exon in the alignment. |
If you used any pairwise alignment method other than Align to Chromosome: DNASTAR, the area will look like the image below.

The columns will include the following information:
| Value | Description |
|---|---|
| % Identity | Number of matching residues divided by the sum of all residues and gap characters. The quotient is multiplied by 100 to give the similarity as a percent. |
| % Gaps | The gap length divided by the consensus length. The quotient is multiplied by 100 to give the similarity as a percent. |
| Identical | Number of residues that are identical in both sequences. |
| Gap Count | Number of gaps (of any length) in either sequence of an alignment. |
| Gap Length | Number of gap characters in an alignment. |
| Score | The cumulative score for an alignment derived by adding the matrix-derived match scores and subtracting gap penalties. |
| Length | Number of residues in the consensus, including gaps that match in both sequences. |
Sequences area:
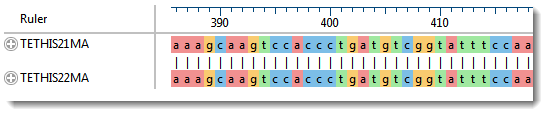
The Sequences area consists of nucleotide or amino acid codes for each sequence on the right, and their labels on the left. The order is always “target (or reference) sequence” first, and “query sequence” second. This section has a different appearance depending on the zoom level. A variety of optional data tracks can be displayed in this section.
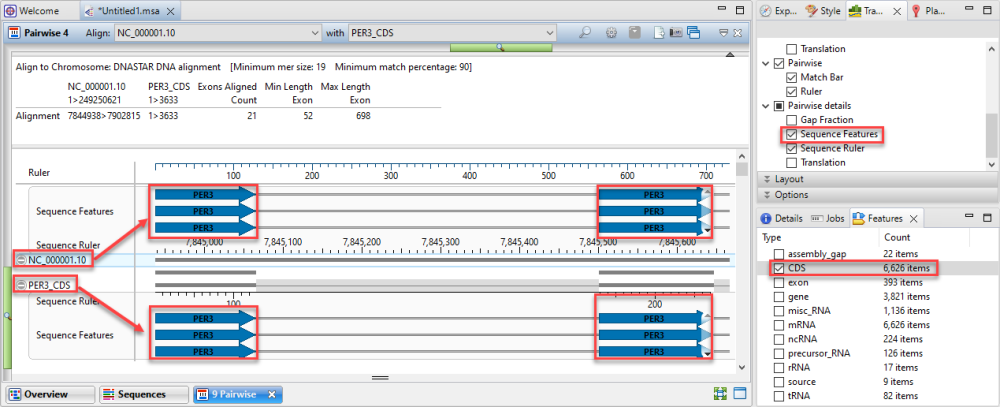
If you used the Align to Chromosome: DNASTAR method, you will likely want to display the Features track for both the reference and query sequences in the Sequences area. You can then use the Features panel to limit the display to only certain types of features, such as CDS features. This lets you visually verify that the features start and end in the same places for both sequences. Features will usually be separated by large stretches of intron sequence.
Tasks pertaining to a Pairwise view that resulted from a pairwise alignment:
In all but one case, the following tasks pertain solely to a Pairwise view that resulted from performing a pairwise alignment. If the Pairwise view resulted from a BLAST search, the only task available is Export image (second row from the bottom).
| Task | How To |
|---|---|
| Customize the look and colors used in the view | Use the Pairwise Alignment section of the Style panel. Available color schemes are described here. |
| Customize the layout of the view | See Customize the look and layout. |
| Copy data from the view to paste into an e-notebook or text editor | First select the range by clicking on the first residue and Shift +clicking on the last residue. Then right-click on the selection and choose Copy. |
| Search for a sequence by name | Click the Search tool ( ) on the right of the header area to open a search area. See Search for a sequence using a text query for details. ) on the right of the header area to open a search area. See Search for a sequence using a text query for details. |
| Change pairwise alignment options | Click the Change alignment options in this view tool ( ), a shortcut to Align > Pairwise. Set options as desired and press Align. ), a shortcut to Align > Pairwise. Set options as desired and press Align. |
| Create a new pairwise subalignment | Select a portion of the alignment, then click the Create new pairwise alignment from selection tool ( ). The subalignment will open in a new Pairwise view. ). The subalignment will open in a new Pairwise view. |
| Export data from the view | Click the Export Data tool ( ), a shortcut to the File > Export Data subcommands. See Export data to a file. ), a shortcut to the File > Export Data subcommands. See Export data to a file. |
| Save an image of the view | Click the Export Image tool ( ), a shortcut to the File > Export Image subcommands. See Export an image of the view. The summary and sequences areas are both exported. ), a shortcut to the File > Export Image subcommands. See Export an image of the view. The summary and sequences areas are both exported. |
| Make a copy of the current view | Click the Clone this view tool ( ). This makes an exact copy of the current Pairwise view, including sequences, alignment type, settings and current selection. This is useful if you want to change one factor for comparison to the original alignment. ). This makes an exact copy of the current Pairwise view, including sequences, alignment type, settings and current selection. This is useful if you want to change one factor for comparison to the original alignment. |
Need more help with this?
Contact DNASTAR