
Now that you have finished Part A of the tutorial, this part shows how you can use a local pairwise alignment to resolve the correct mapping of the first intron in isoF and isoC.
- Click on Pairwise view tab. Look at the left drop-down menu at the top of the view to see that the uppermost sequence (Dmel_ADH) has automatically been selected as the reference.
- In the right drop-down menu, choose "isoF-NM_001022098.2". I The default Local: Smith Waterman method is used to create the alignment
- To make sure all features will be displayed:
- Open the Tracks side-panel.
- Under Pairwise details, click once on the word Features.
- Move the Height slider to the right until you can see all the feature rows.
- Open the Tracks side-panel.
- In the Pairwise view, click on the plus-sign icon next to the name Dmel_ADH to reveal the Features and Sequence Ruler tracks.
A correct alignment would show the 5’ end of the isoF sequence aligned with the two orange arrows that begin at the very start of the Dmel_ADH sequence. Instead, however, the beginnings of both the ADH reference sequence and the mRNA are shown as dimly colored context; in other words, as unaligned flanking sequence.

- Scrolling down through the alignment observe that the 5’ end of the transcript sequence has aligned, albeit poorly, with a region beginning within the first intron. Meanwhile, a solid ungapped alignment doesn’t start until 5’ end of the second exon annotated for either isoF or isoC. (See the boxed area in the image below.)
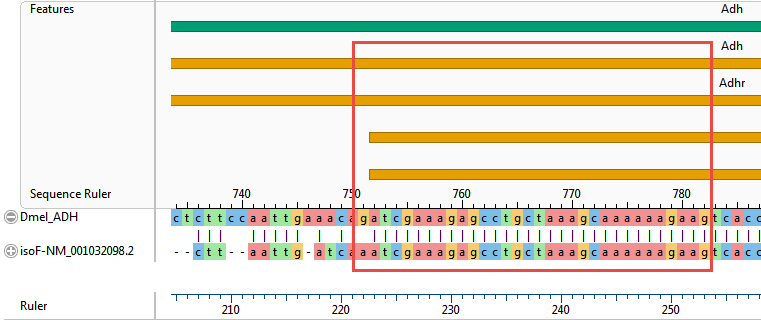
Clearly, a local pairwise alignment was not an improvement over the first two multiple alignments tried in Part A.
Most likely, introducing a sufficient number of gaps to span the intron would reduce the score so much that the aligner can find a higher scoring segment that contains numerous short gapped regions.
Proceed to Part C: Use a Global pairwise alignment method.
Need more help with this?
Contact DNASTAR