
The following instructions describe how to align sequences with similar lengths (i.e., within one or two orders of magnitude). DNASTAR has used these pairwise algorithms in this workflow to successfully align protein sequences up to 35,000 bases in length.
To align two similar-length sequences pairwise:
- Add sequences to the project.
- Decide which two sequences you want to align. A typical workflow is to first perform a multiple alignment, then build a phylogenetic tree, then choose two adjacent sequences to compare.
- From any view, select the two sequences you wish to align. The sequences can vary in length, but both must belong to the same category: DNA/RNA or protein. If one sequence is significantly longer than the other, use drag & drop in the Sequences view or Overview to organize them such that the longer sequence is above the shorter sequence.
- Choose Align > Pairwise or right-click on the selection and choose Align Pairwise to launch the Align Pairwise dialog.
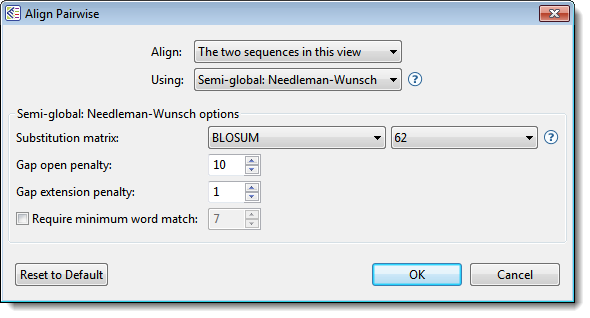
- Choose the desired settings.
- Align – Use this drop-down menu to specify which sequences to align.
- Using – Use this drop-down menu to choose the desired pairwise alignment method: Local, Global, or Semi-Global.
- Substitution matrix – Use this drop-down menu to choose the substitution matrix. A substitution matrix describes the rate at which a nucleotide or amino acid changes to another nucleotide or amino acid over time. Different matrices are available for nucleotide vs. protein sequences, as shown in table below these instructions. Also see the “Notes” section below that for additional information.
- Gap open penalty – Specify the amount that should be deducted from the alignment score for each gap in the alignment. Gaps of different sizes carry the same penalty. Default is 10.
- Gap extension penalty – Specify the amount that should be deducted from the alignment score after first multiplying it by the length of gaps. Longer gaps have a greater penalty than shorter gaps. Default is 1.
- Require minimum word match – If you want to specify the length of the smallest perfect match of contiguous bases/residues to consider in building an alignment, check the box and enter a value. The default is for the box to be unchecked. If checked, the default value is 7.
- Align – Use this drop-down menu to specify which sequences to align.
- Press OK to begin the alignment.
During the alignment, MegAlign Pro displays a progress window. In most cases, this will appear and disappear too suddenly to notice it. In the cases of longer alignments, you can interrupt the alignment, if necessary, by clicking its Cancel button or view a console window showing the start time and progress of the alignment by clicking its Show Console button.
If an alignment finishes successfully, a Pairwise view opens. If an alignment fails, you will receive a message with recommendations on how to obtain a successful alignment (e.g., by modifying options or choosing a different alignment method).
- (optional) If the Console is not already open, and you wish to view alignment statistics and other information there, select View > Console.
Substitution matrix descriptions and options:
| Available for Sequence Type | Matrix | Description | Secondary Option |
|---|---|---|---|
| Nucleotide | NUC44 | DNASTAR’s modified version of NCBI’s NUC.4.4 algorithm, the modification being that U is treated as a synonym of T. In NUC44, exact matches, and T:U matches score as 5, and mismatches between unambiguous bases [G A T C U] score as -4. Matches between bases and ambiguous symbols [S W R Y K M B V H D N] have intermediate scores. A base versus a 2-way ambiguous category [R Y W S K M] to which it belongs scores as +1, and a mismatch to a 2-way group to which it doesn’t belong scores as -4. Example: C is in [S R M] but not in [W Y K] . The 3-way groupings are [B V H D] where C is in all but D (which means not C). Therefore, C vs [B V H] scores as -1 while C vs [D] scores as -4. |
N/A |
| Protein | BLOSUM | (Henikoff & Henikoff, 1992). These matrices are ideal for carrying out similarity searches. | Available matrices range from 30-100, and are provided in increments of 5 and 62. Choose larger numbers for less divergent sequences. |
| Protein | GONNET | Derived from PAM matrices (Dayhoff et al., 1978) but more sensitive, and based on a much larger data set. | (Unchangeable default of 250) |
| Protein | IDENTITY | Scores two identical amino acids as 1, and anything else as -10,000. | N/A |
| Protein | MATCH | Scores two identical amino acids as 1, and anything else as -1. | N/A |
| Protein | PAM | (Dayhoff et al., 1978). Widely used since the late 1970s. | Available matrices range from 10-500, and are provided in increments of 10. Choose larger numbers for more divergent sequences. |
| Protein | VTML | Derived from PAM matrices (Dayhoff et al., 1978) by Müller T et al. (2002), . | Available matrices range from 10-500, and are provided in increments of 10. |
Notes:
- BLOSUM, PAM, GONNET, IDENTITY, and MATCH are part of NCBI’s BLAST distribution. For more information, see NCBI’s substitution matrix page.
- The PAM, GONNET and VTML numbers are based on the presumed millions of years of divergence.
- In BLOSUM, the matrix number is proportional to the presumed degree of similarity. Therefore, BLOSUM100 would be the preferred matrix for near-identical sequences.
- VTLM and GONNET are considered to be updated versions of PAM250.
- In BLOSUM, PAM, and GONNET, match/mismatch scores vary with the series number. Also exact matches vary with the particular amino acid. For example, BLOSUM30 scores W:W as 20 and S:S as 4. BLOSUM100 scores these as 17 and 9, respectively.
Need more help with this?
Contact DNASTAR