
The Assembly Options wizard screen allows you to specify assembly parameters. This screen comes in several variations, two of which are shown below (click on either to see full-size):

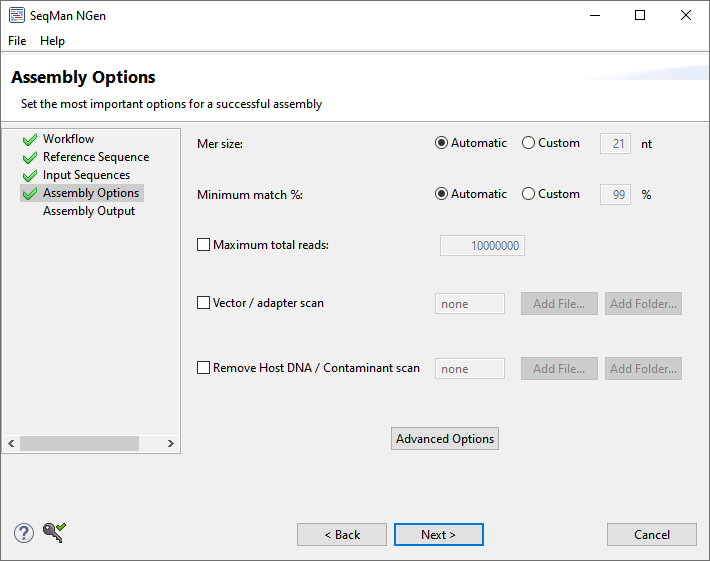
Depending on the workflow, only a subset of the following options will be available:
| Category | Options and Descriptions | |
|---|---|---|
| Coverage calculation | If you know the approximate length of the genome or fragment being assembled, select Estimated genome/contig length and specify a length. SeqMan NGen will then calculate the expected average coverage empirically from the amount of data. This, in turn, allows repeat regions to be identified and handled more accurately, resulting in a better assembly. If the approximate genome length is not known, use the Expected coverage option. If you do not know the length of the genome/fragment, select Estimated coverage and provide an estimate of the depth of the sequencing. The default value for this field is 20, and the maximum allowable value is 65,535. If you enter a value larger than the maximum, you may receive an error message and be prevented from continuing until you choose a value less than or equal to the maximum. Use caution when estimating the value for Expected coverage. If the value you use is significantly lower than the actual depth, the assembly may take a much longer time to complete and may have too many mers flagged as repeats. We recommend using Expected genome length whenever possible. |
|
| Mer size | The minimum length of a mer (overlapping region of a fragment read), in bases, required to be considered a match when arranging reads into contigs. Mer size information is used to identify matches during the assembly layout phase. The default mer size is determined by the selected read technology and is shown in the window. For more information, see How mer tags are chosen.
|
|
| Minimum match % | Specifies the minimum percentage of matches in an overlap required to join two sequences in the same contig. SeqMan NGen determines the percentage to use based on the sequencing technology you specified in the Assembly Options dialog. For more information, see Calculation of.
|
|
| Post assembly options | Check Minimum contig size requirements and type values for one or both of the following to remove assembled, untemplated contigs that do not meet minimum thresholds. This can lead to a desirable decrease in project size.
|
|
| Maximum total reads | Check the box and enter a value if you wish to limit the read depth. Utilizing this option can make the assembly proceed faster. TIP: To see the effect of changes to this parameter, look at the Estimated coverage in the Run Assembly screen. In general, a coverage (AKA “depth”) of 50-100 is ideal and additional depth does nothing but slow the assembly. If your depth differs from the ideal, return to this screen and change Maximum total reads until the Estimated coverage is satisfactory. Note that this value is calculated on a per-assembly basis. If you set up 5 assemblies and set this value to 10 million reads, the cap would be 10 million reads for each assembly, not 2 million for each assembly. |
|
| Vector/adapter scan | Adapter sequences are added to the ends of fragments during sequencing library preparations, and can interfere with downstream processing, if not removed. Check Vector/adapter scan to add either one single- or multiple-sequence .fasta file or one folder of .fasta files containing known or suspected adapter sequences. The file(s) must be in .fasta format. During assembly, sequencing reads will be scanned for the presence of each of the specified adapters and when detected, trimmed off of that read. The trimmed read will then be used in any downstream processing. There is no specific header formatting. There is a minimum exact match length of 11 bases, and a minimum overall match of 15 bases, that allows for some mismatching. Both ends are searched within a specified range (default = 130), and all bases from an identified match to that end of the read are trimmed off. | |
| Remove Host DNA / Contaminant scan | If you want SeqMan NGen to ignore host or contaminant DNA during assembly, check the box and navigate to the file or folder containing the host DNA or contaminant sequence(s). See Remove PhiX control reads from Illumina data. | |
| De novo PacBio/Nanopore options | Choose Assemble all reads or Use subset of reads. If you choose the latter, you may (or must) enter:
|
To set additional assembly options, press the button named Advanced Options or Advanced Assembly Options to open a multi-tabbed dialog. For details on the settings in each tab, see Alignment tab, Layout tab, Trimming tab and Scans tab.
Click Next > to proceed to the next wizard screen or < Back to return to the previous screen.
Need more help with this?
Contact DNASTAR