
If your workflow includes the standard Preassembly Options screen, you can adjust the parameters used for running pre-assembly scans.

In the Read Filtering section.
- To set a threshold for Maximum total reads, check the box and enter the threshold value. The default is for this option to be checked and the threshold to be 10000000. When using Illumina technology, we recommend checking this box and specifying a value to limit the number of reads used in the assembly. For 454 technologies, we suggest unchecking the box and leaving the field blank. If you check this option, be sure to add individual read files rather than folders in the Input Sequences screen. Adding files individually causes SeqMan NGen to use an equal number of reads from each file. If you instead add a folder, SeqMan NGen may potentially use reads from only the first file(s).
TIP: To see the effect of changes to this parameter, look at the Estimated coverage in the Run Assembly screen. In general, a coverage (AKA “depth”) of 50-100 is ideal and additional depth does nothing but slow the assembly. If your depth differs from the ideal, return to this screen and change Maximum total reads until the Estimated coverage is satisfactory.
- To Remove reads smaller than a specified threshold, check the box and enter the threshold value. The default is for this option to be unchecked.
In the End Trimming and Contaminant Scans section, specify whether you would like SeqMan NGen to perform any of the following pre-assembly tasks:
- To automatically trim reads prior to assembly based on quality scores and specified quality end trimming parameters, check Quality end trim.
- To use specified vector/adaptor scan parameters to scan and trim reads for the vector or adapter, check Vector/adapter scan.
- To use specified contaminant scan parameters to scan and remove reads that contain contaminant sequences, check Contaminant scan. If desired, learn how to use Contaminant scan to remove PhiX174 control sequence from Illumina data prior to assembly.
- To use specified repeat scan parameters to scan reads for known repetitive sequences, check Repeat scan. If the option is checked, all sequences identified as repeats will be added to the assembly last, after all non-repeats have been assembled.
If you check any of the last three options, click the Add button to its right to select the desired vector, contaminant, or repetitive sequence(s). This causes a Files and Folders dialog to pop up.
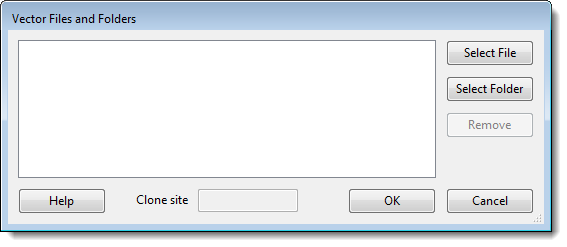
- Select File – Click to navigate to and select one or more individual sequences.
- Select Folder – Click to navigate to and select an entire folder of sequences.
- Clone site (Vector dialog only) – Enter the position of the cloning site where insertion occurs.
- Remove – Click to remove a selected (highlighted) file from the list.
Once you are finished, click OK to save your changes and return to the Preassembly Options dialog.
If desired, click the Advanced Options button to access additional settings. This opens a multi-tabbed dialog. For details, see Trimming tab, Scans tab and Alignment tab.
Click Next > to proceed to the next wizard screen or < Back to return to the previous screen.
Need more help with this?
Contact DNASTAR