
The Alignment tab of the Assembly Options dialog is used to set parameters for the alignment phase of the assembly. To access the tab from the Assembly Options screen, click the Advanced Options button then click on the Alignment tab. The options available in this tab vary depending on the workflow.
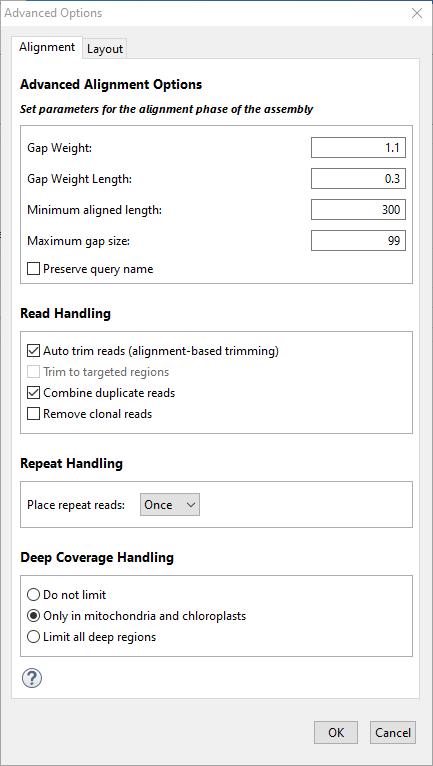
The table below shows editable options in alphabetical order; each workflow includes a subset of these options. Default parameters vary according to the sequencing technology and project type specified elsewhere in the wizard, and values seldom need to be changed.
| Parameter | Description |
|---|---|
| Auto trim reads (alignment-based trimming | If this box is checked, the ends of reads are trimmed to best match alignment to the reference. SeqMan NGen will mark the portion of the read that aligns well to the reference and will set the trimming to skip any of the poorly aligning parts of the read. Checking this option optimizes the end trimming of reads to maintain as much of the read as possible, while still meeting the minimum match percentage threshold. However, checking the box can also lead to the removal of true variant bases located near the ends of reads. The box is checked by default. |
| Combine duplicate reads | Duplicate reads are those which share the same starting position and the same sequence. Check this box if you wish to combine the reads and only enter one of them into the alignment. Any duplicates will be scored but not aligned. Combining duplicate reads collapses reads with identical sequences with the same start and stops and replaces them with a single entry with a suffix “[dup #]” where # is the number of collapsed reads. However, this option does not take the location of a paired end read into consideration. It is used primarily to reduce issues with alignment and visualization of very deep sequence regions, typical of RNA-Seq data for highly expression genes. |
| Deep coverage handling | This section of the Alignment tab dialog lets you specify whether and when to filter deep coverage regions. The default selection is made automatically by SeqMan NGen based on the current workflow. However, you may make any desired selection. The way in which very deep coverage is handled can greatly affect assembly time. For example, if unlimited deep coverage is allowed, it could take upwards of eight hours to align human NA24385 sequences (Genome in a Bottle “Ashkenazim Trio Son”) to the mitochondrial (MT) genome. That is because most of that human sample has a coverage depth of 35,000. In such a case, limiting deep coverage regions can allow the assembly to proceed much more quickly. Choose between:
|
| Gap Weight | The penalty for introducing a gap in a pairwise alignment. |
| Gap Weight Length | The penalty for extending a gap in a pairwise alignment. |
| Layout align | In cases where a read has an identical, or nearly identical, overlap score to more than one location on the reference, indicative of a repeated sequence, the read can be evaluated by attempting a fully gapped alignment to each potential mapping position and selecting the position with the best score. In case of ties, the read is placed in one of the locations at random. Checking this box will further lower the false discovery rate (FDR), but may substantially increase the assembly time. The default is for this box to be unchecked. |
| Layout stringency | To specify the non-permanent “soft” filters for SNP data. SNPs that do not meet thresholds specified in this section are removed from certain displays (e.g., tables) but are still retained in the final project and may be displayed in downstream analysis, if desired. Specify Low, High or Custom stringency. Choosing Custom enables additional options. |
| Maximum gap size | The theoretical maximum length of a gap that could be inserted. In practice, the maximum gap size will usually be about half of this value. The maximum allowable value is 99. |
| Minimum aligned length | The minimum length of at least one aligned segment of a read after trimming. The default value varies depending on the read technology you selected. Allowed values are 0-999. |
| Place repeat reads | Choose to place repeated reads Once, All or Never. The default is All for metagenomics workflow and Once for all other workflows. The way in which very deep coverage is handled can greatly affect assembly time. For example, if unlimited deep coverage is allowed, it could take upwards of eight hours to align human NA24385 sequences (Genome in a Bottle “Ashkenazim Trio Son”) to the mitochondrial (MT) genome. That is because most of that human sample has a coverage depth of 35,000. In such a case, limiting deep coverage regions can allow the assembly to proceed much more quickly. |
| Preserve query name | When SeqMan NGen runs a templated assembly using the XNG assembler, all the sequence read names are normally replaced by “ID” numbers to index the data. Use of this parameter restores the original sequence read name. |
| Remove clonal reads | Clonal reads, where the sequence and endpoints of both reads in a pair match those in another pair, are usually the result of PCR artifacts. Check this box if you wish to retain one of the pairs in the assembly, but completely remove the clones (duplicate pairs) after the alignment phase of assembly. If the box is checked, cloned reads will not be scored, and will not be included in SNP calculation or gene quantification. This option can be useful in genome/exome/gene panel sequencing workflows where clonal reads can skew variant calculations. Checking the box may add substantially to the time required for assembly. Checking this option does not remove a pair if its two reads are duplicates of different pairs. It only removes duplicate pairs if the entire pair is completely identical to another pair. For example, SeqMan NGen will not remove a pair whose forward read is a duplicate of a read from pair A, but whose reverse read is a duplicate of a read from pair B. Note: Do not check both Combine duplicate reads and Remove clonal reads, as this will lead to unpredictable results due to the order in which SeqMan NGen removes clones and combines duplicates. |
| Trim to targeted regions | This box is only enabled for workflows that offer the ability to add a .bed file, and where a .bed file was specified in the Reference Sequence screen If this box is checked, reads extending beyond the 5’ or 3’ end of a targeted region will be trimmed to the target boundary. The box is unchecked by default. |
Once you are finished, click the Layout or Trimming or Scans tabs to change those settings. Or click OK to save all changes and return to the Assembly Options screen.
Need more help with this?
Contact DNASTAR