
This topic pertains to the way SeqMan NGen calculates and GenVision Pro displays structural variations (SVs) for long-read data. This information is partially applicable to Illumina MiSeq data as well.
As of the Lasergene 17.6 release, SV detection in SeqMan NGen relied on split reads for deletion detection and alignment trim edges to detect insertions, with the first method being highly accurate and the second method less so. With the release of Lasergene 18, long reads that can span insertions can also be identified via split reads and detected accurately in terms of location and length.
Long read alignments can resolve insertions that are shorter than the read lengths as shown in the diagram below. SeqMan NGen collects the reads that contains the insertion sequence, de novo assembles them, and creates a structural VCF file that contains the insertion (and flanking region) as one of the output files. In this scenario, a P value is calculated, and the size estimation is usually accurate.
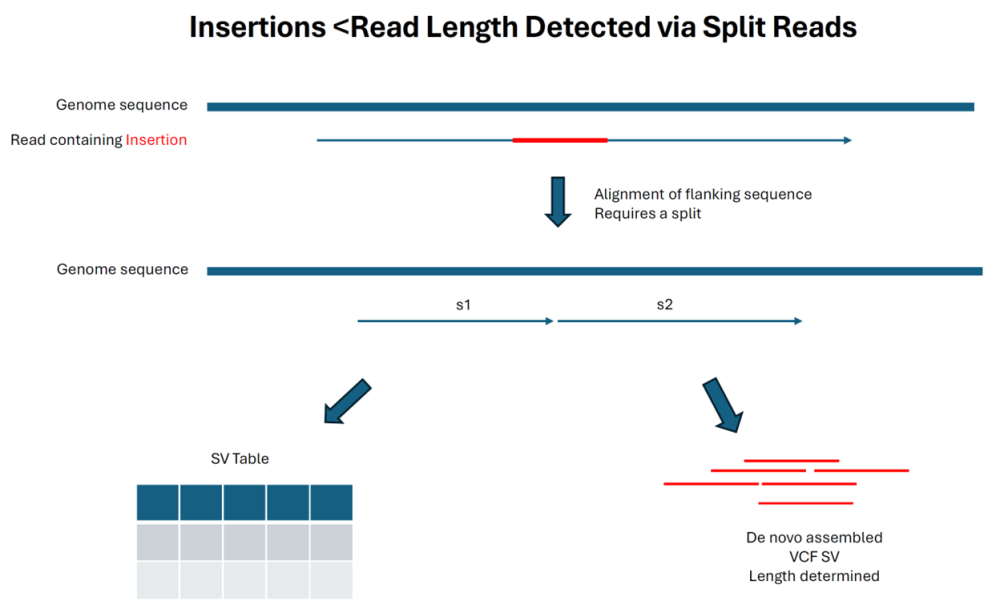
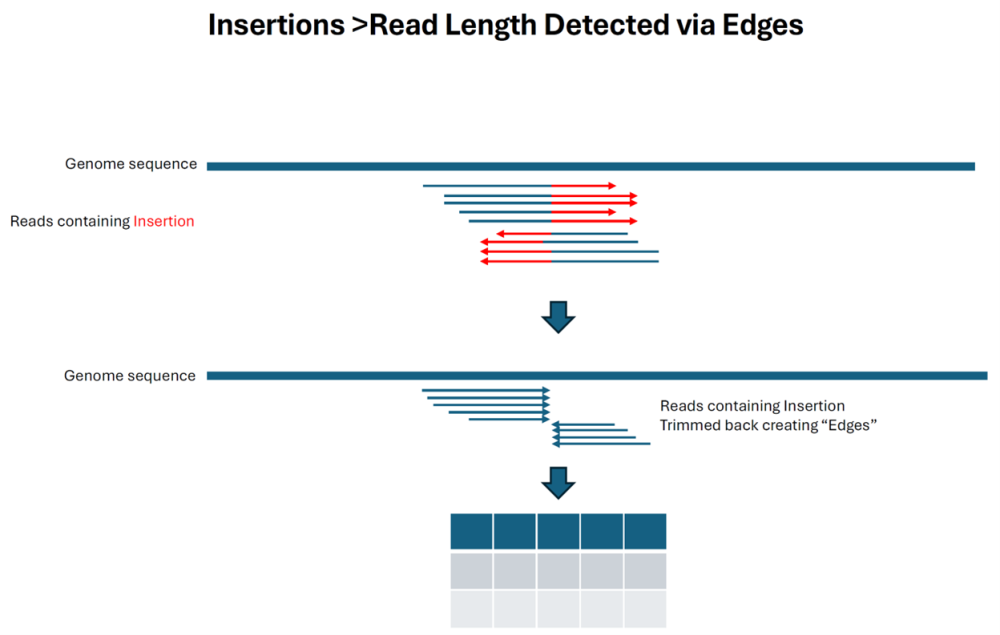
As indicated below, insertions that are longer than the read length are still reported by detecting the trimmed reads flanking the insertion point. Note that this method is much more prone to reporting other alignment artifacts that create trimmed edges as “insertions” and do not have a P value:
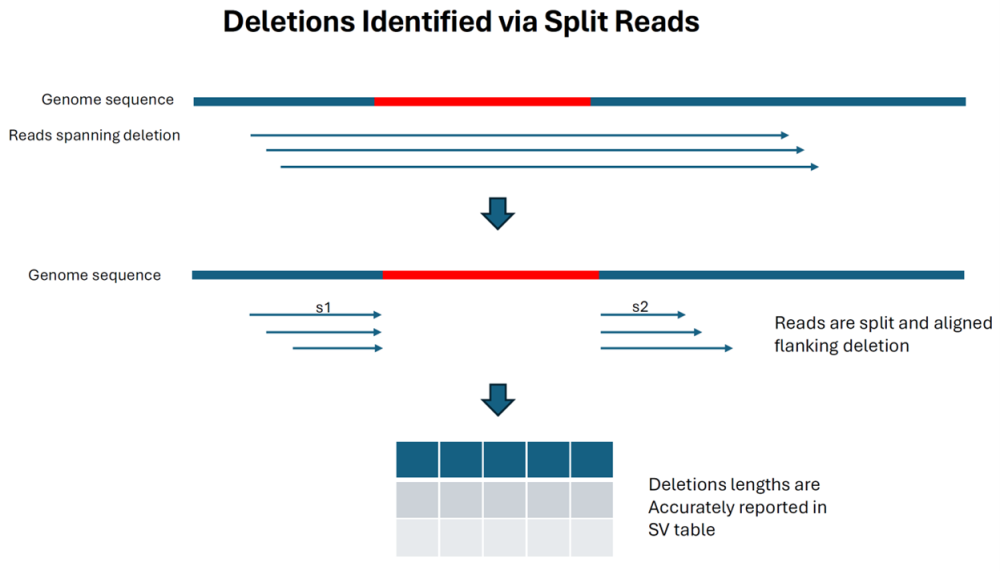
The final diagram shows that deletions are still detected by split reads (as they were in previous versions) and the size limit is not dependent on read length:
Structural variation data for long-read assemblies is saved to VCF SV format.
In-house benchmarking tests:
For benchmarking purposes, we edited an E. coli genome to simulate insertions of 25, 50, 75, 100, 250, 500, 1000 and 2500 bp in length. Deletions were simulated at 25, 100, 1000 and 10000 bp in length. The simulated SVs were placed at regular intervals 50 Kbp apart and a misc_ feature annotation was added at each reference position. This data set highlighted the advantages of the new SV detection algorithm in Lasergene 18. Insertions were detected accurately and length estimation was also accurate.
In the example below, 300 bp Illumina MiSeq reads were aligned to the modified template and insertions up to 75 bp were detected with accurate length estimations. The assembly was opened in GenVision Pro and the Structural Variations table is shown below.
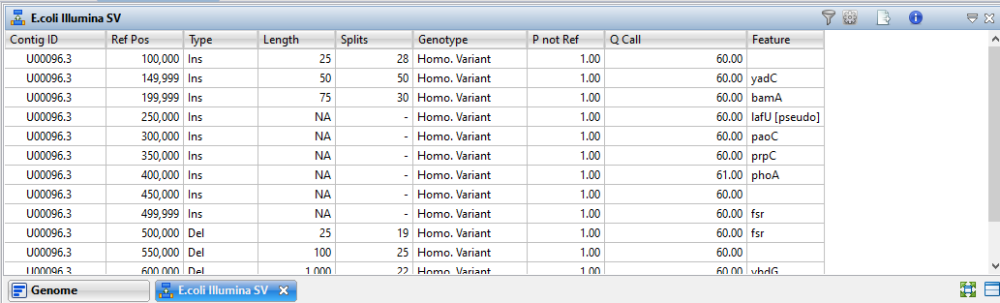
When viewing the results in GenVision Pro, we noted that insertions < 25 bp generally appeared in the Variants view rather than the Structural Variations view. Though length comes into play, if the algorithm can split and align the reads, the finding tends to appear in the Structural Variations view. By contrast, if the algorithm can gap either reads or reference and align the reads, the finding appears in the Variants view.
Aligning PacBio data to the same reference revealed that insertions up to about 1000 bp were detected accurately. With both Illumina and PacBio, larger insertions are detected, but length cannot be estimated.

Need more help with this?
Contact DNASTAR