
The Variant view provides a table with a row for each variant position. This view is only available for reference-guided assemblies in .assembly format.
To access the Variant view, choose a contig from the Explorer panel. Then either choose View > Variants > (Contig Name) or Variants > Show Variant Table or right-click on the selected contig and choose Show Variant Table. In later versions of SeqMan Ultra, you may be able to choose different versions of the view to display using the drop-down menu on the upper left of the view. Currently only available version is SNPs Summary.
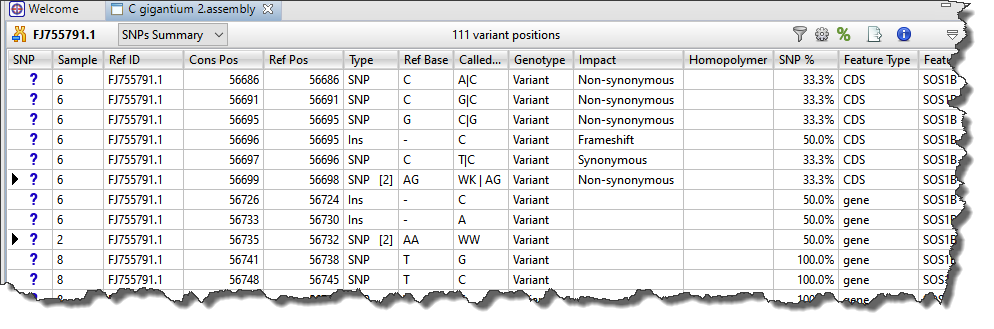
Each row in the SNPs Summary table summarizes information for all of the variant bases in an aligned column. If you are viewing a SeqMan NGen assembly, only variants meeting the SNP filter stringency (High, Medium or Low) that you specified in the SeqMan NGen wizard are displayed.
The following table describes the various columns in this table:
| Column Name | Description |
|---|---|
| SNP | A manual evaluation score for the SNP, with a question mark being the default. See Change the status of a selected variant in the table below for a legend and instructions. |
| Ref ID | The base at this position on the reference chromosome. |
| Cons Pos | The position on a gapped contig corresponding to this chromosome (for SeqMan Ultra and SeqMan NGen assemblies only). |
| Ref Pos | The position on the reference chromosome. |
| Type | Specifies the variation type as SNP, Del (deletion) or Ins (insertion). For .assembly files and certain SQD files (e.g., from de novo or special templated workflows), pink typeface may be used to indicate non-synonymous variants. |
| Ref Base | The base at this position on the reference chromosome. |
| Called Base | The dominant variant in the aligned column. In the case of a heterozygote call, both bases at the position are shown, separated by a vertical bar. For multi-base insertions, the inserted string is shown. For multi-base deletions, the deleted bases are represented with dashes (-). |
| Genotype | Whether this SNP call indicates the reference or not, and (for diploids) whether this calls a homozygous or heterozygous genotype. |
| Impact | The impact of the variant or indel on the genome, displayed as one of the following values:
|
| Homopolymer | Indicates whether the variant occurs within a homopolymeric run, which is defined as two or more identical bases in a row. When using Pacific Biosciences (PacBio) or Ion Torrent data, SeqMan Pro may not list all homopolymeric indels. When possible, insertions or deletions are placed at the 5’ end (top strand) of the run during alignment. |
| P Not Ref | The probability that this position does not match the reference. For combined SNPs and indels, P not ref will be the minimum of the P not refs in the used columns. |
| Q Call | The Phred-like quality score of the called genotype. It is a measure of the confidence that the SNP is present in the sample on a 0-60 log10 scale. For combined SNPs and indels, Q call will be the minimum of all available columns at that reference position. |
| SNP % | The percentage of the sequence at this position in the assembly which varied from the reference. |
| Feature Type | The type of feature as annotated in the template sequence for the gene (e.g. CDS, attenuator, C_region, etc.). |
| Feature Name | If a variant is located within an annotated feature in the reference sequence, the feature type and name are displayed. A single nucleotide change may sometimes be reported as affecting multiple overlapping features. These can include different overlapping genes on the same or opposite strands, as well as alternatively spliced messages from the same gene. In this case, SeqMan NGen produces multiple VCF Variant table entries at the same position, one for each reported feature. A bracketed number follows the Feature Name to indicate which isoform from the Feature view table was used (e.g., TP53 [2]). Note: If a non-gene feature (“mRNA”, “CDS”, etc.) exists in the template file, but has no corresponding “gene” feature, SeqMan NGen adds the “gene” feature automatically during assembly. The locations of any automatically added “gene” annotations are indicated by asterisks (*) in this column. |
| Transcript ID | Transcript ID number from ENSEMBL. |
| DNA Change | The change(s) in the DNA sequence using the nomenclature established by the Human Genome Variation Society (HGVS). |
| Amino Acid Change | The change(s) in the protein sequence using the nomenclature and conventions established by the Human Genome Variation Society (HGVS). The cell text ‘p.(=)’ signifies a synonymous change, while an empty cell denotes a non-coding region. |
| Depth | The number of reads overlapping the aligned column. Since this calculation disregards bases below the quality threshold, the Alignment View may show a greater number of sequences than the Depth shown in the Variants Summary Report. The default quality threshold for assembly in SeqMan NGen is 5. The threshold can be changed either pre-assembly, in SeqMan NGen, or post-assembly, in SeqMan Ultra. |
| Deletion | The number of deleted bases in the Indel. |
| A (C, G, T) Cnt | The number of bases of this type called in the aligned column. A dash (-) represents the reference base. |
The table below describes tasks related to the Variants view:
| Task | How To |
|---|---|
| Open the Alignment view at the variant position | Double-click on any row in the table. This is especially useful in situations such as an amplicon assembled against a much longer reference. In this case, the Alignment view may appear virtually empty when viewed on its own. However, double-clicking on a row in this table will open the Alignment view at a populated position. |
| Select a category of variants | Use the commands in the Variants menu to select a large group of variants at once. Regardless of the current selection, you can select Variants > All or Variants > None. If you have already made a selection in the table, the uppermost menu command will allow you to select all variants of the same type (e.g. Variants > Select > Select All of Type ‘CDS’). To select everything that is not currently selected, use Variants > Invert Selection. Alternatively, you can use the right-click menu options of the same names. |
| Change the status of selected variants | The leftmost column of the table (represented by a small triangle in the header) denotes the status of a variant. Initially, all variants are marked as putative.
|
| Hide/show multiple references bases | If there were multiple reference bases at the location of a variant, the leftmost column will feature a small triangle in that row. Click on the triangle to show the multiple reference bases.  In .assembly projects, variants in adjacent columns are coalesced into a single insertion or deletion if they are of the same type, and if at least 80% of the reads with the called variant in one column have a variant in the adjacent column. Each coalesced multiple-base insertion or deletion can be opened to reveal individual variants by clicking the corresponding triangle to the left of the SNP column. After clicking a triangle, information on each position of the insertion/deletion is displayed in a separate row.  |
| Choose which items should appear in the view | Press the Filter ( ) tool in the top right of the view. See Filtering in the Variants view for detailed information. ) tool in the top right of the view. See Filtering in the Variants view for detailed information. |
| Add, rearrange, rename, or remove table columns | Press the Change options in this view tool ( ) in the top right of the view. For instructions and a description of each column type, see Customize tables in the views. ) in the top right of the view. For instructions and a description of each column type, see Customize tables in the views. |
| Show counts as a percent | Press the Show counts as percent tool (  ) in the top right of the view. This affects columns named Cnt. ) in the top right of the view. This affects columns named Cnt. |
| Export data from the table | Press the Export data tool ( ) in the top right of the view. This opens a save dialog in which you can save the tabular data in comma-separated (.csv) or tab-separated (.tab) format. See Export data from the Variants view for details. ) in the top right of the view. This opens a save dialog in which you can save the tabular data in comma-separated (.csv) or tab-separated (.tab) format. See Export data from the Variants view for details. |
| Toggle between showing/hiding the table header | Use the Show quick details tool ( ) in the top right of the view. An example of the table header appears below boxed in red: ) in the top right of the view. An example of the table header appears below boxed in red:  |
 – Putative (the initial status for all variants in the table)
– Putative (the initial status for all variants in the table)  – Confirmed
– Confirmed  – Rejected
– Rejected
Need more help with this?
Contact DNASTAR