
You can assemble small-to-medium sized Sanger data sets in SeqMan Ultra itself. (Other types of data need to be assembled in SeqMan NGen.)
To assemble small-to-medium sets of Sanger data:
- Do any of the following:
- Click on the Welcome project tab or choose View > Welcome to open the Welcome screen. Click on New Assembly on the left. Then choose New Sanger/ABI assembly from the Molecular biology section.
- Choose File > New Sanger/ABI Assembly.
- Press Ctrl/Cmd+N.
- Click on the Welcome project tab or choose View > Welcome to open the Welcome screen. Click on New Assembly on the left. Then choose New Sanger/ABI assembly from the Molecular biology section.
In each case, the Unassembled view will open.
- In the Unassembled view, upload the sample sequences and, optionally, a reference sequence. To upload sample sequences and their trace data (if available), press the Add button (
![]() ) or choose Edit > Add. To upload reference sequences (optional), press the Add reference button (
) or choose Edit > Add. To upload reference sequences (optional), press the Add reference button (![]() ). In both cases, select the desired files and press Open. Files added using the Add reference button will appear in the table with a checkmark in the Ref column. Sample files that contain trace data will show the word Trace in the Type column.
). In both cases, select the desired files and press Open. Files added using the Add reference button will appear in the table with a checkmark in the Ref column. Sample files that contain trace data will show the word Trace in the Type column.
![]()
p(banner tip). Note: If you try to add large files or non-Sanger sequences, you will receive a message notifying you that SeqMan NGen must be used for the assembly.
![]()
Press OK to launch SeqMan NGen at the Input Sequences wizard screen.
 ) or choose Edit > Add. To upload reference sequences (optional), press the Add reference button (
) or choose Edit > Add. To upload reference sequences (optional), press the Add reference button ( ). In both cases, select the desired files and press Open. Files added using the Add reference button will appear in the table with a checkmark in the Ref column. Sample files that contain trace data will show the word Trace in the Type column.
). In both cases, select the desired files and press Open. Files added using the Add reference button will appear in the table with a checkmark in the Ref column. Sample files that contain trace data will show the word Trace in the Type column. 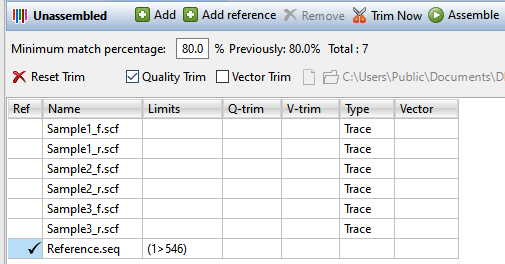

- (optional) If you need to remove rows from the table, select them and press the Remove tool (
![]() ). Prior to removing rows, you can verify how many rows are selected by looking in the middle of the blue header between the two groups of tools.
). Prior to removing rows, you can verify how many rows are selected by looking in the middle of the blue header between the two groups of tools.
![]() .
.
 ). Prior to removing rows, you can verify how many rows are selected by looking in the middle of the blue header between the two groups of tools.
). Prior to removing rows, you can verify how many rows are selected by looking in the middle of the blue header between the two groups of tools.  .
.- (optional) If you wish to perform automatic vector removal, start by checking the Vector Trim box. By default, SeqMan Ultra will remove vector sequence from any of the vectors in the VectorData catalog installed with Lasergene. If desired, you can override this by using the tools to the right of the checkbox. Press Choose a vector from a file to navigate to a specific vector sequence file or Choose a folder containing vectors to specify a folder of vectors. Then press the Trim now tool (
![]() ). If vector sequence was found and removed, the vector name will be listed in the table’s Vector column. The coordinates that will be omitted from the assembly are shown in the V-trim column.
). If vector sequence was found and removed, the vector name will be listed in the table’s Vector column. The coordinates that will be omitted from the assembly are shown in the V-trim column.
 ). If vector sequence was found and removed, the vector name will be listed in the table’s Vector column. The coordinates that will be omitted from the assembly are shown in the V-trim column.
). If vector sequence was found and removed, the vector name will be listed in the table’s Vector column. The coordinates that will be omitted from the assembly are shown in the V-trim column.- (optional) Automatically trim sequence ends based on quality. Start by checking the Quality Trim box and press the Trim now tool (
![]() ). This method uses SeqMan Ultra’s proprietary quality score system to remove low-quality data. The coordinates that will be omitted from the assembly are shown in the Q-trim column of the table above. See Quality score calculations and End-trimming based on averaged quality scores for more information.
). This method uses SeqMan Ultra’s proprietary quality score system to remove low-quality data. The coordinates that will be omitted from the assembly are shown in the Q-trim column of the table above. See Quality score calculations and End-trimming based on averaged quality scores for more information.
- (optional) Manually trim sequence ends. Start by double-clicking on any sample row containing trace data. This populates the lower portion of the Unassembled view with a trace data chromatogram.
![]()
Grab the vertical trim sliders on the 5’ and/or 3’ ends of the sequence and release them in the desired location. The portion of the sequence that will be removed is shown with a yellow background. If you already performed automated quality or vector trimming in the steps above, a trimmed portion may already be visible. You can still use the sliders, if desired, to override the trim location and choose a different one. The coordinates that will be submitted for assembly are shown in the bottom left corner of the Unassembled view, as well as in the Limits column of the table above.
![]()
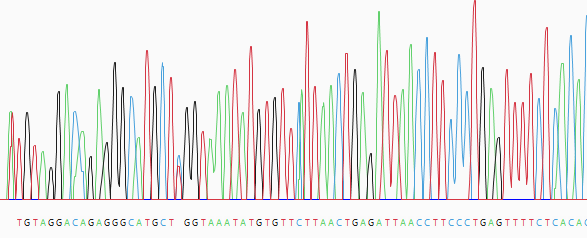
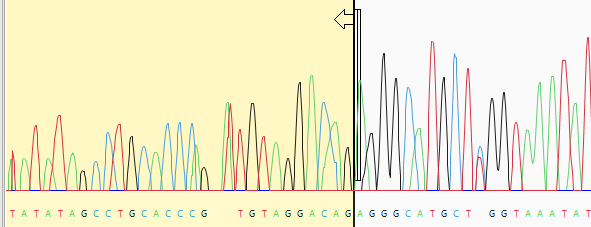
- (optional) If you change your mind and wish to restore all the trimmed sequence, press the Reset Trim tool (
![]() ).
).
 ).
).- Specify the assembly stringency by entering a number in the box to the right of Minimum match percentage in the header. To the right of the box, the header displays the number used the previous time this set was assembled (if an assembly has already taken place) and the number of sample and reference sequences involved in the assembly. If some samples are not being aligned, try changing the Minimum match percentage prior to assembling again.
![]()

- To assemble all the sample sequences in the table, press the Assemble tool (
![]() ). If a reference is present, the assembler will use the reference(s) as a template. Otherwise, the sequences will be de novo assembled.
). If a reference is present, the assembler will use the reference(s) as a template. Otherwise, the sequences will be de novo assembled.
 ). If a reference is present, the assembler will use the reference(s) as a template. Otherwise, the sequences will be de novo assembled.
). If a reference is present, the assembler will use the reference(s) as a template. Otherwise, the sequences will be de novo assembled.The status of the assembly can be checked in the Jobs panel.
Once assembly is complete, results will open in the Project Details view and any contigs will appear in the Explorer panel.
Need more help with this?
Contact DNASTAR