
To select which columns to display in the Variants view, or to rename or reorder them, click the Choose or rearrange columns ( ) tool. The Choose Columns dialog appears.
) tool. The Choose Columns dialog appears.
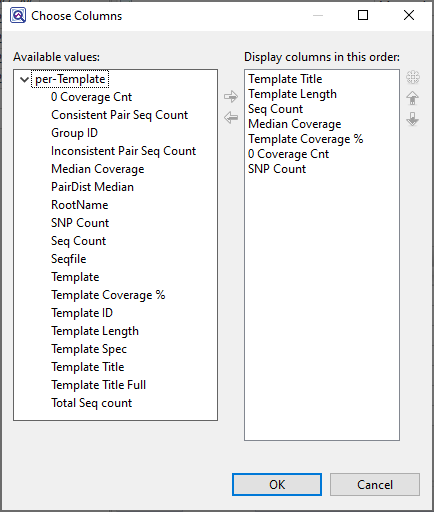
Available columns are on the left, while currently-applied columns are on the right.
- To add a column to the display, select its name on the left and press the right arrow key to move it to the right.
- To remove a column from the display, select its name on the right and press the left arrow key to move it to the left.
- To change the order of displayed columns, select the column name you wish to move on the right, then use the up/down arrows to place it in the desired order.
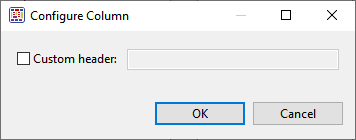
- To create a custom header for a displayed column, select its name on the right, then choose the Configure column tool (
![]() ). In the popup dialog, type in the desired name and press OK.
). In the popup dialog, type in the desired name and press OK.
Descriptions of each column appear below in alphabetical order.
| Column Name | Description |
|---|---|
| Amino Acid Change | This column is only available if Show Codon Bases & Distance to feature is unchecked. The column shows the change(s) in the amino acid sequence, using the nomenclature established by the Human Genome Variation Society and The Sequence Ontology Project. This includes:
|
| Called Base | The dominant variant in the aligned column. In the case of a heterozygote call, both bases at the position are shown, separated by a vertical bar. For multi-base insertions, the inserted string is shown. For multi-base deletions, the deleted bases are represented with dashes (-). |
| Coding Feature Distance | Shows whether variants are within or near a named feature, and the distance from that feature. For .assembly files and certain .sqd files (e.g., from de novo or special templated workflows), the following color scheme may be used:
|
| Codon | When a translated feature is present on the reference sequence at the position of a variant, a codon change is displayed. The codon and amino acid translation is shown for the reference sequence and compared to the codon and amino acid translation for the selected variant. The position number of the amino acid change is also displayed. If more than one translated feature is present at the variant position, SeqMan Ultra will use the first feature based on the current sorting in the Features view. |
| Cons Pos | The position on a gapped contig corresponding to this chromosome (for SeqMan Ultra and SeqMan NGen assemblies only). |
| COSMIC | The Catalogue of Somatic Mutations in Cancer (COSMIC) ID for positions with known variants. Double-clicking on the entry opens the corresponding page at COSMIC. For human assemblies only. |
| Cross Compare | (DNASTAR internal use only) |
| dbSNP ID | The dbSNP rs ID, if available, for positions with known variants. Double-clicking on the entry opens the corresponding page at dbSNP. |
| Deletion | The number of deleted bases in the Indel. |
| Depth | The number of reads overlapping the aligned column. Since this calculation disregards bases below the quality threshold, the Alignment View may show a greater number of sequences than the Depth shown in the Variants Summary Report. The default quality threshold for assembly in SeqMan NGen is 5. The threshold can be changed either pre-assembly, in SeqMan NGen, or post-assembly, in SeqMan Ultra. |
| DNA Change | Change(s) in the DNA sequence affecting either CDS features or splice sites are indicated using the nomenclature established by the Human Genome Variation Society (HGVS). A “c.” prefix, followed by coordinates taken from the ORF, denotes a change in a CDS feature. For example:
|
| Feature Name | If a variant is located within an annotated feature in the reference sequence, the feature type and name are displayed. A single nucleotide change may sometimes be reported as affecting multiple overlapping features. These can include different overlapping genes on the same or opposite strands, as well as alternatively spliced messages from the same gene. In this case, SeqMan NGen produces multiple VCF Variant table entries at the same position, one for each reported feature. A bracketed number follows the Feature Name to indicate which isoform from the Feature view table was used (e.g., TP53 [2]). Note: If a non-gene feature (“mRNA”, “CDS”, etc.) exists in the template file, but has no corresponding “gene” feature, SeqMan NGen adds the “gene” feature automatically during assembly. The locations of any automatically added “gene” annotations are indicated by asterisks (*) in this column. |
| Feature Type | For variants within a gene feature, the feature type is shown in the following order of precedence: CDS, mRNA, Gene. If Show Codon Bases Distance to feature is selected, this column also contains a feature designation if the variant is within 150 bases of the nearest exon. Therefore, it is possible for a variant that is in a gene to also be listed as a CDS, mRNA, etc. When Show Codon Bases is checked, the Feature Type column will also show the distance to the nearby exon and an arrow indicating the direction of the feature.Feature types for different variant locations are shown below:
|
| Genotype | When the “Diploid” SNP detection method is used in a SeqMan NGen assembly, there are four possibilities: 1) homozygous variant (both alleles have the same base and it is different from the reference), 2) reference (both alleles have the same base and it is the same as the reference), 3) heterozygous reference (two different alleles are called, one with the same base as the reference, the other with a variant base), and 4) heterozygous not reference (two different alleles, neither of which match the reference base). It is quite rare for the reference case to occur in the table. This only happens in cases where there is sufficient evidence of the possibility of a variant to pass the filtering threshold, but where the evidence is still quite weak. These cases are usually eliminated by even modest filtering. When the Haploid SNP detection method is used, only variant and reference are possible. Note: In this column, if one or more of the adjacent variants is called as a heterozygote, the coalesced variant is also called a heterozygote. Therefore, for a coalesced variant to be called homozygous, all positions must be called homozygous. |
| GERP | The Genomic Evolutionary Rate Profiling (GERP) score representing the calculated evolutionary constraint at that position. GERP data is automatically delivered when you use DNASTAR’s human template package prior to performing a templated assembly in SeqMan NGen. To limit the size of the data file required, only positions with scores of 1.0 or greater are displayed. GERP is a tool that provides a score for each position in the human genome that estimates whether that position is under purifying selection or not (Davydov et al. 2010). GERP uses alignments between the human genome and 33 other mammalian genomes to quantitate the position-specific constraint in terms of rejected substitutions, defined as the difference between the neutral rate of substitution and the observed rate, estimated by maximum likelihood. Substitutions in sites under selection are assumed to be more deleterious than those not under selection. Scores range from negative values to ~6. Positions with scores below or near zero are not under selection. Conversely, the more positive the score, the more constrained the position. GERP information can be useful in evaluating the impact of non-synonymous variants in coding regions and the impact of changes in or near promoter elements, among others. |
| Homopolymer | Indicates whether the variant occurs within a homopolymeric run, which is defined as two or more identical bases in a row. When using Pacific Biosciences (PacBio) or Ion Torrent data, SeqMan Pro may not list all homopolymeric indels. When possible, insertions or deletions are placed at the 5’ end (top strand) of the run during alignment. |
| Impact | The impact of the variant or indel on the genome, displayed as one of the following values:
|
| PDB ID | Worldwide Protein Data Bank (PDB) ID number. |
| P Not Ref | The probability that this position does not match the reference. For combined SNPs and indels, P not ref will be the minimum of the P not refs in the used columns. |
| Q Call | The Phred-like quality score of the called genotype. It is a measure of the confidence that the SNP is present in the sample on a 0-60 log10 scale. For combined SNPs and indels, Q call will be the minimum of all available columns at that reference position. |
| Ref Base | The reference sequence base in this position. For multi-base deletions, the reference sequence of the string is shown, beginning with the base at the Ref Pos coordinate. If there is no reference sequence present, the Ref Base column displays the most frequently occurring non-ambiguous base at this position. If no such base exists, the consensus base at this position is shown. |
| Ref ID | Reference sequence or chromosome. |
| Ref Pos | Reference position that does not include gaps. Coordinates matching entries in the VCF Variant table are shown. For deletions, Ref Pos is the genomic coordinate of the first deleted base. For insertions, Ref Pos is the genomic coordinate of the base preceding the insertion. |
| Region Capture | Indicates whether the variant occurs within a region specified in the .bed or manifest file used. Values are Yes and No. |
| Residue Count (A Cnt, etc.) | The number of bases of this type called in the aligned column. A dash (-) represents the reference base. |
| Sample | The sample name. |
| SNP | A manual evaluation score for the SNP, with a question mark being the default. See Change the status of a selected variant in the table below for a legend and instructions. |
| SNP % | The percentage of the sequence at this position in the assembly which varied from the reference. |
| Splice | Variant is in or near an exon splice site. Splice site variations are changes to the 5’ (“donor”) or 3’ (“acceptor”) consensus splice site sequences. The DNA sequence for the donor is 5’-AGGTRAGT-3’ and for the acceptor is 5’-YYYYYYYYCAGGT-3’. Note that the AG dinucleotide on the 5’ end of the donor and the GT dinucleotide on the 3’ end of the acceptor are within the exon. Therefore, changes at these positions can also cause changes in the amino acid sequence of the resulting proteins. Changes in the intron portion of the splice site are marked as “Splice” in the column while those in the exon portion of the site are labeled “Splice in CDS.” Only the position where the change occurs is considered, not the identity of the base. |
| Trace% | (DNASTAR internal use only) |
| Transcript ID | Transcript ID number from ENSEMBL. |
| Type | Specifies the variation type as SNP, Del (deletion) or Ins (insertion). For .assembly files and certain SQD files (e.g., from de novo or special templated workflows), pink typeface may be used to indicate non-synonymous variants. |
| User ID | Positions corresponding to a custom VCF Variant Table are labeled with the ID from that set. |

Need more help with this?
Contact DNASTAR