
The Extract Features as Sequences template, located in the Templates panel, prompts you to choose the feature of interest, to enter an annotated sequence and a sequence range, and to choose the desired file output and location. Later, when you run the script, SeqNinja will extract the features from the sequence and output the results to the selected file.
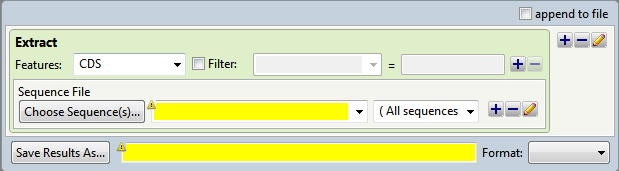
- In the Features area, select the feature type of interest from the drop-down menu, or type its name in the menu box. If you want to extract more than one feature type to the same output file, use the plus (+) button to add additional feature type lines. If you want to further limit the search to features matching particular qualifiers, check the Filter box, then choose a qualifier from the drop-down menu to its right. In the right-most textbox, enter the text that the qualifier must match (e.g., /gene = thrL). You may use wildcards in this box if you wish (e.g., /gene = thr*).
- In the Choose Sequence(s) button row, choose the annotated sequence from which you wish to extract features (see Add and modify a sequence).
- In the Save Results As area, choose the name and location in which to save the output (see Specify output format and location).
Within the output file, SeqNinja automatically creates individual sequence names by extracting feature information in the priority order shown below. If the first piece of information is not available, SeqNinja attempts to name based on the next item on the lis,t and so on. In order, SeqNinja attempts to create names based on:
- the qualifiers gene or locus_tag
- a general set of preferred qualifiers
- any qualifier with a value
- the feature key plus an index (for example, CDS:1). Note that if you attempt to extract features of type DNA, with no qualifier values, the sequence names may simply consist of: “DNA:1”, “DNA:2,” etc.
Example:
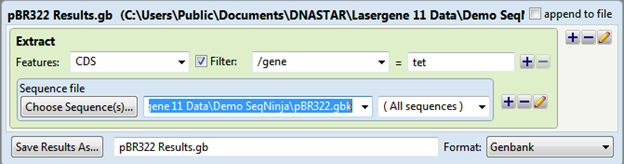
The output is saved as pBR322 Results.gb and the file contents are shown below:
> SYNPBR322:tet
ATGAAATCTAACAATGCGCTCA…
Need more help with this?
Contact DNASTAR