
TIPS FOR SUCCESSFUL DE NOVO TRANSCRIPTOME SEQUENCE ASSEMBLY
USING RNA-SEQ DATA
DOWNLOAD THIS GUIDE
Chapter 1: The De Novo Transcriptome Analysis Workflow
What is a transcriptome?
The functions of most genes in living organisms are still unknown. Human DNA, for example, contains over 20,000 genes, but only ten percent of them have been studied in detail. While each cell of an organism contains the same genes, only a subset of these genes is switched on, or “expressed” in a given cell.
Genes that are currently being expressed are constantly transcribed (copied) into pieces of mRNA. The “transcripts” (copies) travel to the cell’s ribosome, where they serve as instructions for making proteins. The collection of all transcripts in a tissue or cell type is called a “transcriptome.”
Transcriptome analysis is one way to learn more about gene function in a particular tissue or cell type. By analyzing the transcriptome in an experimental tissue sample, we can determine its “gene expression,” including which proteins were being produced at the time the tissue sample was collected. As our knowledge of different transcriptomes becomes more complete, we are learning how tissue cells normally function and how changes in the transcriptome may reflect or contribute to disease. In addition, knowledge about gene expression provides vital clues about the functions of different genes.
To download this entire ebook as a PDF, click here.
What are the two main ways to assemble transcriptome
read data?
During laboratory preparation for sample sequencing, nucleotide chains are broken into thousands of shorter fragments. Bioinformatics software is later used to reassemble those fragments into the longest chains possible. The software does this by automatically finding which reads partially overlap and using that information to reassemble the sequence fragments into longer sequences called “contigs.”
Until relatively recently, most transcriptome data was assembled by mapping it against a reference genome. This type of assembly is known as a “templated” assembly and involves uploading both the experimental reads and a (usually annotated) reference sequence into the assembly software. The short sequence fragments are then aligned to the reference sequence template. This type of assembly is fast, efficient, and creates long contigs that can be automatically annotated using the information from the reference sequence.
By contrast, “de novo” assembly utilizes only the experimental reads; no reference sequence is involved. This type of assembly results in numerous short contigs, takes longer to do, and requires a much larger amount of computing power. In addition, the lack of a reference sequence normally means that de novo assembled contigs cannot be annotated.
Why assemble reads de novo instead of using a whole-genome reference sequence as a template?
Given the disadvantages of de novo assemblies, why wouldn’t everyone elect to do templated assemblies instead? The most common reason is that many organisms do not have a reference sequence available.
But even if a reference sequence is available, templated assemblies have several disadvantages that may make de novo assembly more desirable.
- Transcriptome coverage is usually correlated with levels of gene expression in a particular cell or tissue type. By contrast, genome sequence coverage levels can vary randomly due to repeated content in intron regions of DNA.
- Reference-guided assembly can’t account for alternative splicing and other types of structural alterations of mRNA transcripts.
- Spliced variants that did not align continuously along the genome might be erroneously written off as protein isoforms.
- If the reference genome was incomplete, that could lead to only a partial recovery of transcripts that would have been found using de novo assembly.
The second part of this ebook will discuss a software solution that eliminates many of the downsides of de novo assembly and allows contigs to be annotated for both model and non-model organisms.
What are the steps involved in de novo transcriptome assembly and analysis?
Depending on your project, objective, and the applications you are using, de novo transcriptome assembly and analysis projects can require a variable number of steps. In this ebook, we have divided the bioinformatics portion of the workflow into four steps based on when you would typically need to switch from one tool to the next to keep advancing through the pipeline.
If you are using open-source solutions, there may be additional steps involved that are not discussed here. Conversely, if you are using certain commercial software, multiple steps may take place in a single process. Whichever route you take, it’s important to preplan your software pipeline, even before you send your samples for sequencing.

Preliminary Steps: Prepare samples and obtain RNA-Seq data
The workflow begins in your laboratory when you extract and purify mRNA from tissue in your organism of interest.
The samples are then sequenced in-house or sent to a sequencing facility. Sequencing reverse-transcribes the mRNA and creates a complementary DNA (cDNA) library which is then fragmented into shorter lengths. The most common sequencing technologies for this workflow are Illumina MiSeq and HiSeq paired reads, and data will usually be provided in the FASTQ file format.
When obtaining reads for use in the de novo transcriptome workflow, you should aim for the longest reads possible. You might be tempted to de novo assemble your 500 million, 2x76bp read Illumina RNA-Seq data set. However, these assembled transcripts will be truncated (shorter) compared to an assembly run using longer reads.
While 50-100bp paired Illumina reads are perfectly suitable for templated RNA-seq analysis, de novo transcriptome assembly is greatly improved when using paired Illumina reads that are 150bp or greater in length (Figure 1 and the yellow rows in Table 1).
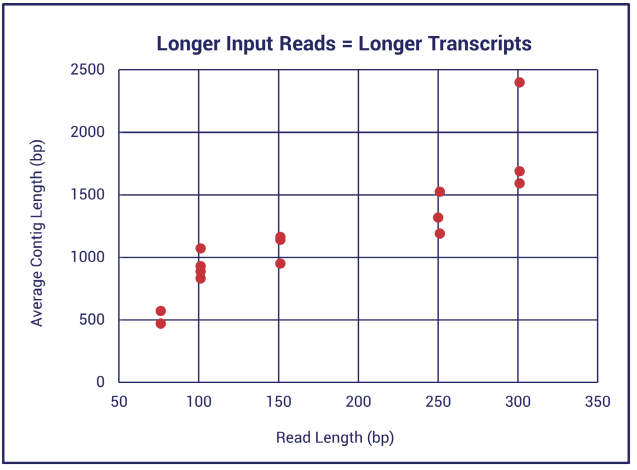
Figure 1. Contig lengths obtained in in de novo transcriptome assemblies for fifteen different organisms using five different Illumina paired read lengths.
Illumina reads over 150bp in length typically produce much longer assembled transcripts–up to full length. Meanwhile, reads less than 150bp may produce transcripts as little as half the length of the mRNA. Assembly of reads that are too short will produce many partial transcripts.
Next, assembly software will be used to assemble the cDNA sequence reads into transcripts. In some cases, the assembly software can also remove vector and adapter sequence prior to assembly and annotate transcripts during assembly. In other cases, you will have to use different software applications to do each of these. The order of the steps below is a typical order followed when using open-source software.
Software Step 1: Find and remove contaminants, vectors, and adapters
Transcriptome sequencing data commonly contains contaminants and untrimmed adapters (AKA “linkers” or “vectors”) that severely impair de novo assembly. For example, over half the transcriptome data sets that we have downloaded from the National Center for Biotechnology Information (NCBI) Short Read Archive contain what we consider to be unacceptably high levels of the “Illumina Universal Adapter.”
Prior to assembly, you can use FASTQC (free download Babraham Institute) to scan your .fastq input data files for the presence of adapters. If your files contain adapter sequence, look for assembly software that can automatically remove it prior to assembly (Figure 2). After removing adapter sequence, data sets assemble in a fraction of the time, with longer and more completely assembled mRNA transcripts.

Figure 2. SeqMan NGen assembly software provides a checkbox to remove universal adapter, or to remove user-specified vectors and adapters.
Software Step 2: Assemble the transcripts
No matter what assembly software you use, it will prompt you to upload your cDNA reads, generally in FASTQ format. Some software allows you to optimize assembly settings. To begin the assembly, you usually just click a button. Depending on your data set, computer hardware, and the assembly software you are using, de novo transcriptome assembly can take from an hour to a week to finish.
Assembly of large transcriptome data sets usually takes at least several days on a desktop computer and for this reason, these data sets are often assembled on computer clusters.

Why can’t I just do a whole genome assembly with my RNA-Seq data?
Why not just do a whole genome assembly with your data? There are at least three main reasons why we recommend instead following a transcriptome-specific workflow.
First, high sequence coverage in whole genome assembly may be the result of repetitive sequences, which can be hidden during assembly. However, when analyzing transcriptome data, high sequence coverage is more likely to indicate abundance, which is important to know about.
Next, RNA-Seq can be strand-specific because both sense and antisense transcripts may be present. By contrast, genome sequencing always uses both strands. And finally, transcript variants from the same gene are hard to resolve using whole genome assembly because they can share exons.
Software Step 3: Annotate the transcripts
If you are using typical assembly software, especially open-source software, de novo assembly of RNA-Seq data can result in thousands of unlabeled contigs representing the expressed transcripts.
Functional annotation of the assembled transcripts provides information about biological processes and molecular functions of specific proteins. Some software can perform functional annotation of the assembled transcripts as part of the assembly process. If not, transcript annotation takes place once assembly is complete.
BLAST-based annotation approaches can be extremely time consuming and difficult to manage. One of these solutions is the subscription-based Blast2GO (B2G), which mines Gene Ontology website data. Another is GOanna, an open-source, agriculture-centric solution that is part of the AgBase database. Data sets containing thousands of query sequences can take multiple days to annotate using these approaches.
Software Step 4: Analyze the transcripts
Once assembly is finished, results are usually displayed in a table. If the assembly software provided an auto-annotation option, the transcripts may be sorted into “identified” and “novel” categories. The novel category includes all transcripts that did not match items in the annotation database selected in the assembly application. Some software may allow you to sort the transcripts using filtering or sorting functions, and to export a selected set of transcripts.
More commonly, the table will simply present thousands of contigs representing the expressed transcripts, without any context or labels. In this case, novel transcripts can sometimes be identified using additional annotation options like DNA to protein database matching.
What are common challenges in this workflow?
De novo assembly of whole transcriptome sequencing data from non-model organisms presents some unique challenges, starting with the sequencing data itself.
The number of reads in the typical de novo transcriptome assembly is vast and can consist of up to half a billion reads. The presence of short Illumina reads, untrimmed Illumina adapter (linker) sequences, ribosomal RNA, and/or genomic repetitive sequences often results in failed or poor quality assemblies. Large transcriptome data sets often require computers with more RAM and CPU power than is readily available in most desktop computers.
Finding a workable software pipeline presents another problem, especially if you are limited to open-source solutions.

In order to produce a complete and annotated mRNA set from the contig assembly, you will likely need to master multiple assembly and annotation applications. For example, the output from a typical sequence assembly program can include thousands of unannotated contigs that have to be identified and annotated using additional software tools.
But while de novo transcriptome assemblies can be challenging, these issues can be managed with careful thought and preparation.