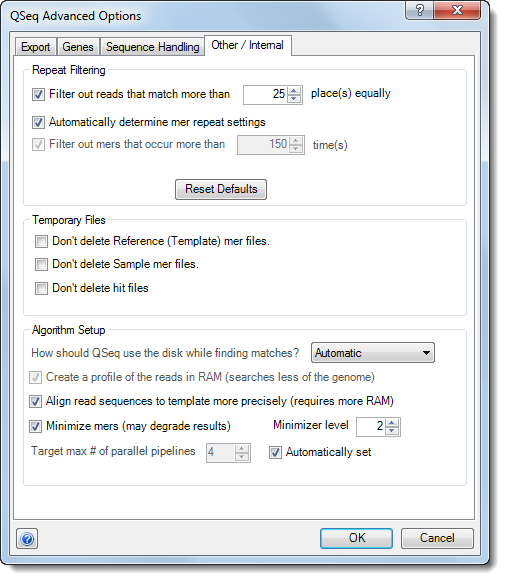
•In Repeat Filtering section, specify how to handle mers that are repeated in the template sequence(s). Choose from:
o Filter out reads that match more than n place(s) equally – Mers that match more than the specified number of places in the template equally well will be excluded from processing.
o Automatically determine mer repeat settings – QSeq automatically determines which mers to include or exclude from processing.
o Filter out mers that occur more than n times – Reads containing mers that occur more than the specified number of times will be excluded from the processing.
•Reads that are not filtered out by these options and that meet the read assignment and mer length requirements will be mapped to the template.
•Click the Reset Defaults button if you would like to revert to the factory default settings.
•The options in the Temporary Files section allow you to keep the temporary files that are created during the QSeq processing if you desire. Selecting any of these options may significantly increase the amount of disk space used and are primarily reserved for troubleshooting purposes. In general, it is not necessary to select any of these options unless directed to do so by a DNASTAR representative.
•Use the drop-down menu under Algorithm Setup to specify the level of memory and disk space to be used when processing your QSeq data. Choose from Automatic, Mixed Usage, Force RAM Usage, or Force Disk Usage In general, Automatic will give the best performance, but under certain conditions, changing this setting may optimize data processing. Please contact support@dnastar.com for more information about changing this setting.
If desired, check any of the following boxes:
•Create a profile of the reads in RAM – Check this box to use a profile of the target read data that ignores irrelevant portions of the genome to which reads cannot map. Checking the box may be useful:
o In cases where reads map to a small portion of the genome, such as the gene regions for RNA-Seq or the binding sites for ChIP-Seq.
o When ChIP-Seq data contains many 'noise' reads which cause the read profile to be unmanageable.
o As an alternative to data-paring options which may bias data by ignoring alternate layouts for the reads occurring elsewhere in the genome.
o However, in cases where reads cover most of the genome, or anytime RAM allows (e.g. a human sized genome and a computer with 16 GB RAM), we recommend leaving this option unchecked.
•Align read sequences to template more precisely – This performs an approximation of a full-read sequence alignment on reads, once their possible locations are determined using mer matches, in order to find more precise matches. In particular, checking this option compensates for substitution errors in short reads, near the ends of reads, and strings of substitution errors spaced near one another—all of which can cause problems for mer-based-only layout. If this box is unchecked, QSeq only uses mers of the length chosen. If you uncheck the box, we recommend lowering should the percentage under Read Assignment Qualification Options to allow for less precise handling of base calling errors and other mismatches (i.e. SNPs).
•Minimize mers – When checked, the layout is performed using pattern matching to thin the mers, thus improving efficiency. Checking this option does, however, increase the chance of a read being unassigned or placed incorrectly. This option requires you to select a Minimizer level. Minimizer levels of 1-2 are generally “safe,” and will cause very few reads to be lost or misplaced, with the exception of very short reads. With long reads of high quality, level 3 is also relatively “safe.” Unchecking Minimize mers or reducing the Minimizer level will slow QSeq and increase the minimum RAM requirements of the application.
•Automatically set – Check this option to let QSeq choose the optimal number of pipelines to run in parallel on different processors/cores. By default, it may attempt to create up to one pipeline per processor core it detects on the computer. To override the default setting, uncheck the box and enter a lower value next to Target max # of parallel pipelines.