
To open the Variant Filter Criteria dialog from the All Found Variants report or the Variants Summary report click the Filter button near the left of the report header.
For BAM-based assemblies (.assembly), the Variant Filter Criteria dialog contains a larger number of options. The exact options present depend on a variety of factors (e.g., the assembly options chosen in SeqMan NGen).
For SeqMan Pro (.sqd) files, the Variants Filter Criteria is abbreviated.
Each item in the Variant Filter Criteria dialogs is optional, including checkboxes, drop-down menus and text boxes. You may check or uncheck any combination of checkboxes to filter for particular types of variants. You can enter information into the text boxes or clear every text box to use the SeqMan Pro defaults.
Variant Filter Criteria dialog changes are applied immediately to the Variant Report. Settings are automatically saved every time you close the dialog and are preserved even if you close and later reopen the project. Items in this dialog act as “soft” filters, meaning that they limit which variants appear in SeqMan Pro reports without actually removing data from the project.
The following table contains detailed information about each section of the Variant Filter Criteria dialog. As noted above, many of these options are not available for SeqMan Pro (.sqd) projects.
|
Section |
Item Name |
Description |
|
Type |
Substitution |
Check this box to filter for substitutions, the locations where one amino acid replaces another amino acid. |
|
Indel |
Check this box to filter for indels, short insertions or deletions. Optionally, you may enter a minimum size for these indels using the Min Size text box. | |
|
Genotype |
For assemblies using the diploid variant caller, use the drop-down menu to filter for variants that are Homozygous only, Heterozygous only, or Any (either type). This option is not available for assemblies using the heterogeneous variant caller. | |
|
Functional Impact |
Not coding |
Check this box to filter for variants that appear in non-coding regions. |
|
Max distance to coding feature |
Check this box to filter for variants occurring within the specified distance from an exon or CDS feature. For example, entering “100” will limit variants to those located 100 bases or less from a feature of the type “exon” or “CDS.” Allowable values are 0-250. The distance from each variant to a coding feature is displayed in the Coding Feature Distance column in the Variants Summary report. Note that data are only displayed if the variant is within a gene. | |
|
Synonymous |
Check this box to filter for variants that do not alter the translated amino acid sequence. | |
|
Non-Synonymous |
Check Non-Synonymous to filter for variants that alter the translated amino acid sequence. Checking this box automatically checks all types below it. The box can also be unchecked, and the boxes below it checked individually:
•Substitution filters only for variants causing single amino acid substitutions.
•In-Frame Indel filters for indels that are evenly divisible by three.
•No-start filters for changes resulting in the absence of a start codon.
•No-stop filters for changes resulting in the absence of a stop codon.
•Nonsense filters for changes resulting in a premature stop codon.
•Frameshift filter for changes caused by indels that are not evenly divisible by three. | |
|
Splice Sites
|
Use the drop-down menu to filter for variants that are Inside Splice Sites Only, Outside Splice Sites Only, or Anywhere. | |
|
Alignment |
P Not Ref |
For assemblies using the diploid or haploid variant caller, enter a number to specify the minimum probability that the genotype at this position is not the reference genotype. The default value depends on the chosen variant filter stringency: High is 90%, Medium is 75%, and Low is 50%. The minimum allowed value is 10%. The P Not Ref option is not available for assemblies using the heterogeneous variant caller. |
|
Q-call |
For assemblies using the diploid or haploid variant caller, enter a number to specify the minimum confidence score. This option is not available for assemblies using the heterogeneous variant caller. | |
|
SNP Percent |
Enter numbers in the boxes to display only those variants occurring within the specified percentage range. For example, entering 50 in the min(imum) field and 90 in the max(imum) field will update the report to only display variants that occur in at least 50% but no greater than 90% of the bases for that position. By default, this filter is set to display all variants (0-100%). However, if your project contains over 100,000 sequences, SeqMan Pro automatically sets the minimum value to 1% to improve the speed of processing. | |
|
Depth |
Enter numbers in the boxes to filter out variants that do not meet the specified min(imum) or max(imum) depth of coverage.
Note: Depth filtering is applied to NGS data but not to Sanger reads when viewing Sanger Validation results. Because there are typically only a small number of Sanger reads, any Sanger-called variants will remain in the Variant Reports, including those with depths of “one.” | |
|
Include Homopolymer length discrepancies |
A homopolymer is a string of identical bases, such as CCC or TTTTT. Check this box to include variants that result from length discrepancies in a homopolymer region or uncheck to hide those variants. | |
|
Databases |
dbSNP, userSNP, COSMIC |
Use the drop-down menus to choose between:
•Annotated SNPs only – show only the variants that are included in the given database.
•Novel SNPs only – show only the variants that are missing from the given database.
•Any – show all variants, whether or not they occur in the given database (default). |
|
GERP Score |
Show only the variants with a GERP score above the number provided. You must type the number into the box provided. | |
|
In Targeted Regions Only |
Check this box to hide variants outside of targeted regions in the .bed or manifest file specified in the SeqMan NGen assembly. This box should be checked if you are performing a capture assembly. | |
|
|
Restore Project Defaults |
Click this button if you would like to return the Variant Filter Criteria dialog to the default settings. |
Note: Unchecking all boxes in the Type or Functional Impact sections will hide all variants in the Variant Report.
The information in the report header is updated automatically as the filters change:
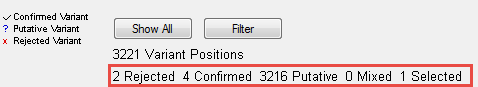
Note: The “Mixed” and “Filtered” categories are currently disabled. Any number displayed for those categories should be ignored.
While the Variant Filter Criteria dialog is open, you can use the File > Print menu command to print a simple list of the options in the dialog, along with a header indicating the date and the project name.