The MACS Peak Finder is based on the peak detection algorithm (Zhang, et al., 2008). This is a model-based algorithm that expects there to be paired peaks of reads on either side of a true binding site. The algorithm attempts to build a model of the distance between these peaks and takes this distance into account to shift reads forwards or backwards, resulting in a peak centered over the true binding site.
The MACS Peak Detection algorithm reports the number of reads within a peak as the signal value for that peak. It also calculates a P-value based on the distribution of reads near a peak region to try to compensate for uneven background noise across the genome. When present, control data are used to filter the peaks that are called and to assign each peak an FDR score which is the false discovery rate likelihood that the peak is not valid.
Note: Control data can only be used for ChIP-Seq experiments. You may specify control experiments in the Create Binding Proteins dialog of the Project Setup Wizard.
MACS reports the start and end position for each peak. It also reports the position of the highest point within the peak and the number of reads that map to that position. These can be accessed through the "Start", "End", "Pinnacle", and "Top Height" annotation values in the Peak Table after assembly.
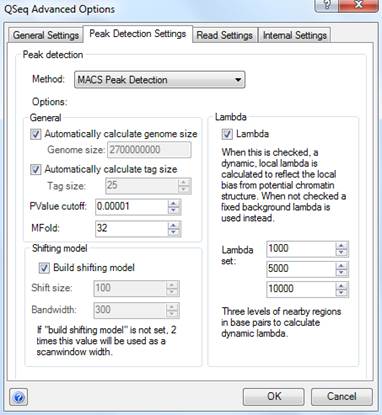
If you choose MACS Peak Detection as the peak detection method, the following options will be available:
•In the General settings section, you may specify a Genome size or have QSeq automatically calculate the genome size based on the template sequences you have loaded. QSeq uses the genome size to calculate how many reads are needed to call a region a peak during the model building stage. The expected distribution is based on the total number of reads and the total size of the genome. QSeq treats all reads as if they were of equal length, defined by the Tag Size. As with genome size, you may specify a tag size manually or have QSeq automatically calculate the average read length and use this value as the tag size. QSeq will calculate the likelihood that a detected peak is actually a peak based on the local read distribution and only return peaks with values below the PValue cutoff. By default, this value is set to 0.00001. The MFold parameter is used during the model building to control how enriched a peak has to be against the background read distribution to be considered in building the peak model.
•In the Shifting model settings section, either check Build Shifting Model to have MACS build a model based on the data to determine the width of and the distance between the "paired peaks," or leave this option unchecked to set Shift Size and Bandwidth values manually. The Shift Size is the distance each of the paired peaks will be shifted to try to center them over the actual binding site. The Bandwidth value defines the expected width of peaks. QSeq will search for peaks using a window twice as long as the bandwidth.
•The Lambda parameter is used to define a Poisson distribution which MACS uses to determine the expected number of reads in a given region. When the Lambda option is checked, the Poisson distribution is calculated for the peak region and for three regions surrounding the peak. The maximum lambda of those regions is used when deciding whether to call a region a peak. If this option is unchecked, then the local distributions are not calculated and instead the expected distribution is based on the total number of reads and the effective size of the genome. The Lambda set value determines the size of the three regions on either side of the peak, in bases, that will be used when calculating the three local distributions.