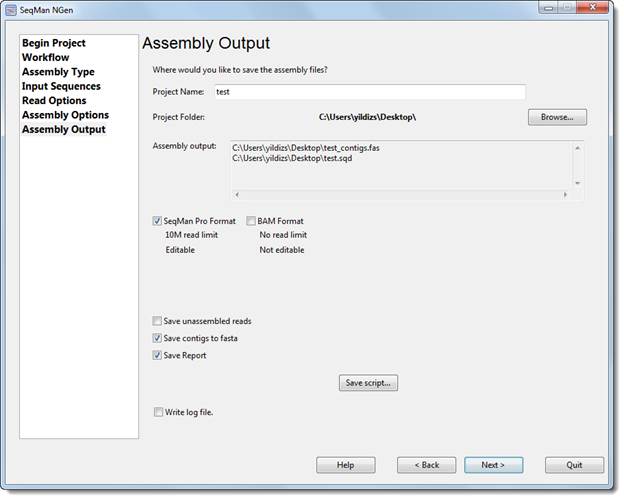
You must select a name and location for your project in the Assembly Output dialog before proceeding further in the wizard. The following version of the dialog is shown only when you are following the de novo or special reference-guided workflows.
Note: For other workflows, see Assembly Output (All Others).
There are two mandatory fields in this dialog:
•Project name – Enter a name for all output files, including the finished assembly.
•Project folder – Use the Browse button to select a location for your assembly output files.
o For non-Cloud assemblies, Browse launches your file explorer. Navigate to the desired location and then click Open to exit. The required disk space may range from 1 GB to 5 TB, depending on a variety of factors. See our technical requirements page for more information.
Note: Never save the assembly output files directly to the desktop, as the many intermediate files and folders created during assembly may hamper or prevent further computer operations. However, files may be saved to a folder on the desktop.
o For Cloud assemblies,
Browse opens the DNASTAR Cloud Data Drive and displays your files on the
DNASTAR Cloud. Navigate to the desired location and highlight the target folder,
then click the green check mark ( ) to exit from the DNASTAR Cloud Data
Drive.
) to exit from the DNASTAR Cloud Data
Drive.
The Assembly output display shows the output file extensions based on current workflow and other selections.
|
Workflow |
Default assembly output |
|
.sqd and .assembly | |
|
De novo Transcriptome/RNA-Seq (transcript annotation workflow) |
.transcriptome |
|
Other de novo |
_contigs.fas and .sqd are always present; .assembly depends checkboxes below (if available)
_unasm.fastq is present if the Save unassembled reads checkbox is checked.
.assembly is present if BAM Format is checked. |
|
All others |
.assembly is always present; .sqd depends checkboxes below (if available); .log is present if Write log file is checked. |
The following checkboxes let you request additional output files. These will all have the name and location specified above, but different file extensions:
•SeqMan Pro Format –Check this box to save assembly output files in both .assembly format and .sqd format; both will be saved in the same location. A DNASTAR .sqd file is editable in SeqMan Pro, but is limited in size to 10 million assembled reads. For projects with more than 10 million reads, no .sqd will be produced even if this option is selected.
•BAM Format - Check to save assembly output files in DNASTAR’s .assembly format, which has a no read limit. This selection lets you view, but not edit, the finished assembly in SeqMan Pro.
•Save unassembled reads – Check the box to save all sequences that were not assembled in the project as a multi-sequence .fastq file. If desired, you may use SeqMan Pro to query the BLAST database to determine why these particular sequences did not fit into the assembly. A default quality score of 15 will be given to each base.
•Save contigs to fasta – Check the box to save the consensus sequences from each contig in the assembly as a multi-sequence .fasta file.
•Save Report – Check the box to save an assembly report text file. If the SeqMan Pro Format checkbox is checked, all report information is saved within the SeqMan Project file (.sqd), even if you do not check this box. To view the report in SeqMan Pro, choose Project > Report.
Note for Windows users: To open a text report with the correct formatting displayed, we recommend using Wordpad, Notepad++, or Microsoft Excel®, and not the default Windows text editor, Notepad.
•Save Script – Press this button if you wish to save your project and convert your wizard choices into a SeqMan NGen assembly script (.script) prior to assembly. (A copy of the script is saved automatically when you initiate an assembly from the “Your assembly is ready to begin” screen.) This button is not available for Cloud assemblies.
The resulting assembly script is an editable text file that can be modified and re-run if desired. To do this, start a new project and specify Rerun analysis of existing assemblies in the Choose Assembly Workflow screen.
Note: If you use Save Script after having checked the Run as separate assemblies box in the Input Sequence Files and Define Experiments or Individual Replicates screen, a set of three separate scripts will be saved for the project. If you save one or more of these scripts to a location other than the main project folder, any attempt to run the assemblies from the SeqMan NGen project script will fail. Moving the projects back to the main project folder will allow assembly to proceed.
•Write log file – If this box is checked (as it is by default in version 14.1 and later), SeqMan NGen will output a complete log file of the assembly process in text format. The file is saved in the same project folder as the .assembly and is assigned the name [project_name].log. The log includes the SeqMan NGen script for the project, followed by a list of steps that were performed and their outcomes.

This log is especially useful in troubleshooting an assembly that will not complete.
Once you are finished, click Next > to continue to the next wizard screen.
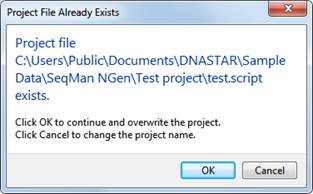
Note: If you choose a name that already exists in the chosen location, you will receive the following warning. Click OK to continue and over-write the earlier project; or Cancel to return to the wizard screen, where you may change the project name and/or location.